Marfan症候群および類縁疾患Marfan Syndrome and Related Disorders
東京大学医学部附属病院小児科Department of Pediatrics, The University of Tokyo Hospital ◇ Tokyo, Japan
発行日:2025年2月28日Published: February 28, 2025
Marfan類縁疾患とは,Marfan症候群と類似した症状や特徴を持つ遺伝性結合組織疾患を指し,Marfan症候群とともに遺伝性大動脈疾患(heritable thoracic aortic disease: HTAD)に含有される.近年の遺伝子解析の進歩により,これらの疾患の関連遺伝子が多く明らかになり,診断および重症度分類における遺伝子検査の重要性が増している.Marfan症候群は一生涯にわたって進行する多系統疾患で,遺伝科・循環器科・眼科・整形外科・胸部外科・産科の専門科チームと連携しながら,生涯にわたってフォローをする必要がある.また,年齢によって注意すべき症状が異なる.特に大動脈基部拡張の評価・治療が重要で,内科的・外科的治療の選択肢を踏まえながらフォローする必要がある.また,妊娠リスクに関する理解など,小児期から成人移行を見据えた管理が必要である.
Marfan syndrome is a genetic disorder that affects the connective tissue. Many conditions share features with Marfan syndrome, and they are referred to as heritable thoracic aortic diseases. Recent advances in genetic analysis have revealed many genes associated with these disorders, indicating the importance of genetic testing in diagnosis and severity classification. Marfan syndrome is a multisystem disorder that progresses over a lifetime and requires lifelong follow-up with a team of specialists in genetic medicine, cardiology, ophthalmology, orthopedics, thoracic surgery, and obstetrics. Additionally, the symptoms to watch for vary with age. The evaluation and treatment of aortic root dilation are particularly important, and follow-up should be based on the medical and surgical treatment options. Moreover, management should focus on the patients’ transition from childhood to adulthood, including an understanding of pregnancy risk.
Key words: heritable thoracic aortic disease; multisystem disorder; annulo-aortic ectasia; pregnancy; transition
© 2025 特定非営利活動法人日本小児循環器学会© 2025 Japanese Society of Pediatric Cardiology and Cardiac Surgery
Marfan類縁疾患とは,Marfan症候群と類似した症状や特徴を持つ遺伝性結合組織疾患を指し,Marfan症候群とともに遺伝性大動脈疾患(heritable thoracic aortic disease: HTAD)に含有される.HTADは,胸部大動脈瘤以外の多臓器の病変を伴うsyndromic HTADと胸部大動脈瘤以外の明確な身体的特徴を伴わないnonsyndromic HTADに分けられ,Marfan症候群とその類縁疾患はsyndromic HTADに含まれる.Marfan症候群との鑑別が重要な症候群として,Loeys–Dietz症候群,血管型Ehlers–Danlos症候群,先天性拘縮性クモ指症,Shprintzen–Goldberg症候群などが挙げられる.近年の遺伝子解析の進歩により,これらの疾患の関連遺伝子が多く明らかになってきた(Table 1)1).
| 疾患名 | 原因遺伝子 | 遺伝形式 | 主な特徴 | その他の特徴 |
|---|---|---|---|---|
| 非症候群性遺伝性胸部大動脈瘤・解離 | ACTA2, MYH11, MYLK, PRKG1, LOX, MAT2A, MFAP5, FBN1, TGFB2, TGFBR1, TGFBR2, SMAD3 | AD | 胸部大動脈瘤・解離が主症状 | 遺伝子によって異なる |
| 症候群性遺伝性大動脈疾患 | ||||
| 高頻度に大動脈病変を伴うもの | ||||
| Marfan症候群 | FBN1 | AD | 大動脈瘤,僧帽弁疾患,水晶体偏位,骨格異常 | 自然気胸 |
| Loeys–Dietz症候群 | TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3, SMAD2 | AD | 大動脈瘤・解離,動脈蛇行,口蓋裂,二分口蓋垂 | 心臓弁膜症 |
| 血管型Ehlers–Danlos症候群 | COL3A1 | AD | 全身の動脈瘤・解離,消化管破裂,頸動脈海綿静脈洞瘻 | 気胸,薄い皮膚,小関節過可動 |
| 動脈蛇行症候群 | SLC2A10 | AR | 動脈蛇行 | 肺動脈,大動脈,その他の動脈の狭窄,大動脈瘤 |
| 時に大動脈病変を伴うもの | ||||
| EFEMP2関連皮膚弛緩症 | EFEMP2 | AR | 皮膚弛緩,動脈蛇行 | 大動脈瘤,肺動脈と大動脈峡部の狭窄 |
| Meester–Loeys症候群 | BGN | XL | 骨格異常 | 大動脈瘤・解離 |
| ホモシスチン尿症 | CBS | AR | 水晶体変異,Marfan症候群様体型,神経障害,血栓塞栓症 | 大動脈瘤・解離 |
| Alport症候群 | COL4A5 | XL | 腎疾患,聴力障害,眼異常 | 胸腹部大動脈瘤 |
| ELN関連皮膚弛緩症 | ELN | AD | 皮膚弛緩 | 大動脈基部拡張 |
| Beals症候群 | FBN2 | AD | 多発性関節拘縮・耳介変形・長い指 | 大動脈基部拡張 |
| FLNA異常症 | FLNA | XL | 脳室周囲結節性異所性灰白質,血小板機能異常,肺高血圧,先天性心疾患 | 大動脈基部拡張,心臓弁異常 |
| Shprintzen–Goldberg症候群 | SKI | AD | 精神運動発達遅滞,頭蓋顔面異常,骨格異常 | 大動脈基部拡張,僧帽弁異常 |
| AD: 常染色体顕性遺伝,AR: 常染色体潜性遺伝,XL: X連鎖遺伝. | ||||
Marfan症候群は幅広い表現型を持つ全身性の結合組織疾患であり,遺伝科・循環器科・眼科・整形外科・胸部外科・産科の専門科チームと連携しながら,生涯にわたってフォローをする必要がある.Marfan症候群の診断は,家族歴の聴取・大動脈基部拡張の評価(小児科・循環器科)・水晶体偏位の評価(眼科)・骨格の基準の評価(整形外科)・遺伝学的検査(遺伝科)など2010年に発表された改訂ゲント基準に基づいて行う(Fig. 1)2).2010年の改訂ゲント基準は,それまでの診断基準と比べて,大動脈拡張と水晶体偏位を重視し簡便化したことと,FBN1変異の位置づけを明確化したことに特徴がある.Marfan症候群が疑われる患者に対する検査・診断の流れをFig. 2に示す.近年は,HTADに対する遺伝子解析を保険診療で行うことが可能になっており,かずさ遺伝子検査室などで関連遺伝子群の受託検査を行っている.

Ao(Z≥2)=大動脈基部拡張,EL=水晶体偏位,FBN1=フィブリリン1遺伝子異常,Syst(≥7 pts)=骨格基準7ポイント以上.
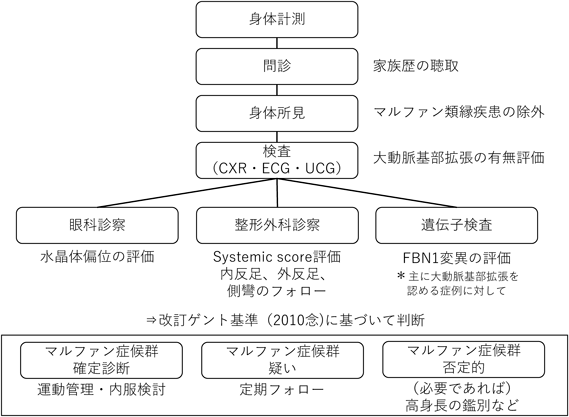
小児期にMarfan症候群の診断基準を満たさなくても,将来的にMarfan症候群や遺伝性大動脈疾患と診断される可能性が否定できないため,大動脈基部が大きめだが正常範囲(Z value<2)の症例などでは慎重にフォローを行う.小児のMarfan症候群の患者では,表現型だけでは,診断基準を満たさない例があり,早期診断のために遺伝子検査が特に有用である.特に,新生児(または乳児)Marfan症候群はMarfan症候群の最重症型と考えられているにもかかわらず,Marfan症候群としての確定診断は難しい.新生児Marfan症候群では,FBN1の遺伝子解析により,新生児領域と呼ばれる(GRCh38リファレンスで)エクソン25-33の変異が見つかることが多い.また,関連遺伝子のコピー数多型がsyndromic HTADの原因となっている症例もあり,通常の変異解析で変異が見つからない場合はコピー数多型解析が診断に有用なことがある3).
近年はFBN1遺伝子の表現型と遺伝子型の関係が明らかになり,患者の遺伝子変異を同定することでMarfan症候群における様々な症状の発症時期の予測が可能になってきた.例えば,蛋白質のジスルフィド結合に関与するシステイン残基における変異では水晶体偏位との関連が知られており,未熟終始コドンを生じて遺伝子産物の不足が起こる変異(ハプロ不全群)では大動脈病変の進行が報告されている4).一方で,当院では,従来遅発型と考えられていたミスセンス変異群(Dominant Negative群)の中にも,大動脈基部置換手術や大動脈解離などの大動脈イベント発生年齢が早い変異型を持つ一群(DN-CD群:エクソン25-36, 43-49内でのCysteine残基に関連したミスセンス変異またはエクソン欠損Deletion)を新たに見出した.Fig. 3に示すように,DN-CD群(28名)は,早発型であるハプロ不全群と同等(30.0[95%信頼区間:19.0–42.0]歳)に早期に血管イベントを発症していた5).また,側弯症の進行に関しても,ハプロ不全群やエクソン25-33のミスセンス変異が重症化のリスク因子になることが報告されている6).
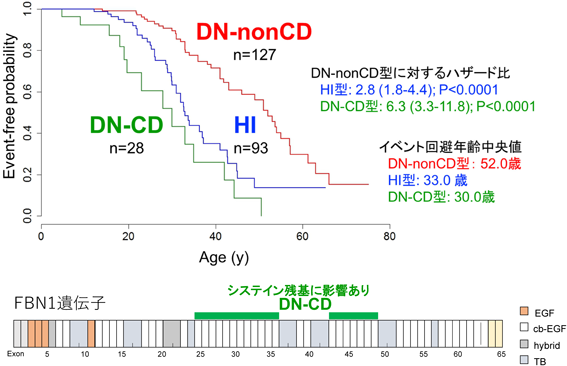
DN-CD, dominant negative variants affecting or creating cysteine residues and in-frame deletion variants in the central cb-EGF domains; DN-nonCD, dominant negative variants not affecting or creating cysteine residues and in-frame deletion variants in the central cb-EGF domains; HI, haploinsufficiency. 文献5より改変して引用.
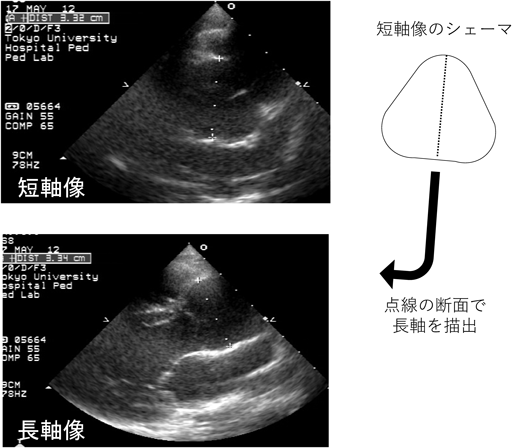
Marfan症候群の診断および治療方針の決定には,心臓超音波を用いた大動脈基部拡張の評価が重要である.Valsalva洞径のZ value≥2を大動脈基部拡張ありと判断するが,改訂ゲント基準においては,Z value≥3が家族歴のある小児における基準になっていることに注意する.小児においては,成長に伴い大動脈基部径の絶対値が増加することが想定されるため,経時的変化はZ valueで比較する.小児においては様々なValsalva洞径のZ valueの計測方法・計算式がある.改訂ゲント基準でも,米国マルファン財団のホームページに記載されている収縮期のInner-edge法やRomanらの報告にある拡張期のLeading edge法の両方が記載されているが,当院では後者を用いている(Fig. 4)7).診断だけでなく治療適応を検討する際にも,Romanらと同様の方法で大動脈基部径を計測するのが一般的だが,この方法は験者間のばらつきが比較的大きい方法であるため,特に経時的変化を評価する際には注意が必要である.Fig. 5に示すように,多くのMarfan症候群の患者において,大動脈基部の短軸像は円形というより三角形に近い形状をしている.そのため筆者は,短軸像で三角形の高さにあたる部分の距離を計測した後に,その短軸で計測した方向の断面を長軸で描出し,大動脈基部径の計測を行っている.正しい断面の描出ができている場合は,短軸と長軸の計測径は一致し,再現性高く大動脈基部の径を評価することができる.Romanらの報告のように,心室の長軸にあわせて大動脈基部を描出した場合,Fig. 4のシェーマにあるようにValsalva洞は胸壁側も背側も膨らんで見えるが,Fig. 5の方法では背側のValsalva洞はほぼ直線状に描出される.小児のMarfan症候群で,経時的変化を評価する際は,再現性の高いFig. 5の方法が有用である.小児期に大動脈弁閉鎖不全を認めることはまれであるが,特に基部拡張が進行した例では注意する.最終的に手術適応を評価する際は,造影CTで動脈瘤の大きさの評価を行うが,その際は動脈瘤の最大短径の計測を行う8).
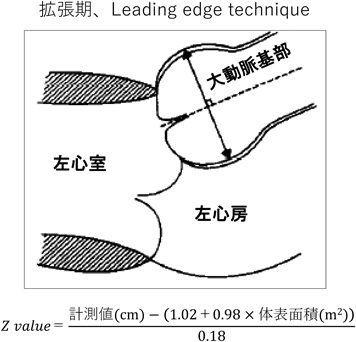
大動脈基部径の計測方法およびZ valueの計算式の一例を示す.
僧帽弁逸脱はMarfan症候群の約30%に認めるとされ,小児でも中等度以上の僧帽弁閉鎖不全症を認めることがある.僧帽弁逸脱による僧帽弁閉鎖不全症の場合逆流は収縮後期に限局されていることが多く,カラードップラーのジェット面積からの評価は重症度を過大評価する傾向がある.そのため,左房・左心室の拡大の程度,肺静脈の血流パターンなどを参考に総合的に重症度の判断,悪化の有無を判断する.一方で,収縮後期に限局されていた僧帽弁逆流が汎収縮期になったときは悪化の兆候であるため注意する.
弁逆流の程度と無関係に心機能の低下を来す症例もあるため,心機能の評価も同時に行う.漏斗胸がある場合は右心系の圧迫を起こしている場合があるので,これについても評価が必要である.
当院では,状態の安定している場合は年に一度,治療介入を検討している場合は半年に一度の頻度で心臓超音波による評価を行っている.
Marfan症候群の眼症状には,水晶体偏位,近視,網膜剥離などがある.水晶体偏位があっても,視力低下の訴えがない場合があるので,無症状の場合でもMarfan症候群が疑われる患者では,積極的に眼科診察を勧める.水晶体偏位は幼少時早期に現れることが多いが,10代で発症する場合もあるため,年に一回は眼科診察を行う.弱視の予防のため,水晶体偏位の早期発見と矯正が必要である.
整形外科への紹介が必要な骨格症状もあり,整形外科との連携も重要である.側弯症はMarfan症候群の患者の約6割に認められ,腰痛・背部痛,拘束性換気障害などを起こすことがあり,装具やGrowing rodを用いた脊柱固定術のタイミングについて相談する.また偏平足があると,足底や膝関節の痛みにつながるが,足底板により症状緩和が得られることもある.漏斗胸は重症化すると,拘束性換気障害を起こしたり,心臓を圧迫し循環に影響を与えたりすることがある.身長が伸びはじめる思春期にかけて急激に陥凹が進行することがあるので注意が必要である.陥凹に伴う症状がある場合や,美容上の理由で手術適応が決まるが,胸腔鏡補助下に金属製のプレートを胸の陥凹部の側面より挿入し,陥凹部を挙上する術式(Nuss法)が広く導入されている.ただしペクタスバー留置中は開胸手術が行えないので,Nuss手術施行前には心臓手術の必要性の評価を行う必要がある.開胸手術を行う予定のある患者では,開胸手術に合わせてRavitch法による胸骨挙上も検討される.
自然気胸はMarfan症候群の患者の4~11%に発生し,思春期以降に問題となることが多い.Marfan症候群における胸痛の重要な鑑別診断であり,特に思春期以降は気胸が起こった時の対応などについて指導する.無症状であることもあるので,胸部レントゲンを撮影した際は,気胸やブラの有無に注意する.
Loeys–Dietz症候群では,脳動脈瘤のリスクもあるため,定期的に頭部MRIを施行することが推奨されている9).
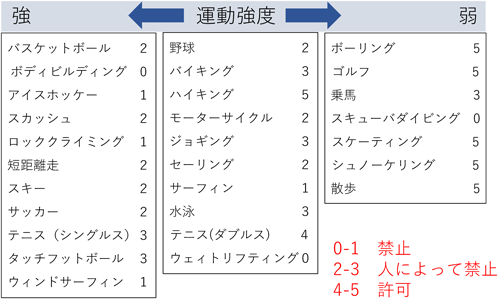
小児Marfan症候群における運動制限の考え方については,2004年のアメリカ心臓病学会からの身体活動に関する提言10)が参考になる(Fig. 6).この提言ではスポーツの種類ごとに推奨レベルが記載されているが,実際にこの通りに細かく指導を行うことは外来診療では難しい.当院では,大動脈基部拡張のある患者,解離の家族歴のあるMarfan症候群の患者では,①ウェイトリフティングなどのアイソメトリックエキササイズ,②高度のエアロビックエキササイズ,③身体衝突の可能性のあるスポーツ,については避けるように指導を行っている.学校生活管理指導表の区分としては,大動脈基部径などにより患者ごとに調整・相談が必要だが,当院ではMarfan症候群と診断されている場合は小学校高学年からD管理かつコンタクトスポーツ禁止としていることが多い.
Marfan症候群ではベータ遮断薬やアンギオテンシン受容体拮抗薬による大動脈基部の拡張抑制効果が報告されている.当院では,Marfan症候群と診断された患者に対しては,副作用の可能性などについて説明したうえで,①経過観察のみ,②Losartan内服,③Losartan, Athenolol併用のどれかを保護者に選択してもらっている11, 12).Losartanは0.5 mg/kg/dayから開始し,1.4 mg/kg/day(上限100 mg/day)まで漸増,Athenololは,0.5 mg/kg/dayから開始し,2 mg/kg/day(上限100 mg/day)まで漸増する.Loeys–Dietz症候群においても,Marfan症候群に準じて内服治療を行う.当院では,患者の多くがLosartan単剤による治療を選択しているが,Losartan開始時の平均年齢は12歳,平均大動脈基部径は約30 mmである.Losartan開始時は立ちくらみや頭痛などを訴える場合もあるが,副作用のために薬物治療を継続できない症例はほとんどない.患者の多くが無症状で,薬剤内服による効果を感じることはないため,思春期になると服薬コンプライアンスを保つのが難しいため,薬物治療の目的について繰り返し説明する.
小児Marfan症候群における大動脈基部置換の適応に関して確立された基準はない.Hannover Medical SchoolのOnoらの報告13)では,大動脈基部径が正常値の200%を超える場合を手術適応としており,German Heart Centre MunichのLangeらは大動脈基部径のZ valueが5を超える場合を手術適応として提唱している14).一方で,12歳未満のMarfan症候群では大動脈が解離することは非常にまれであり,大動脈基部径のZ valueを手術適応の指標にする必要はないという意見もある.そうした視点から,Johns Hopkins HospitalのPatelらは,小児であっても成人の手術適応に準ずるという立場に立っている15).当院では,小児に対しても弁温存術(David手術)を第一選択としているが,大動脈弁の変形が進行する前に手術を行う必要があり,大動脈弁閉鎖不全の進行度も手術適応を決めるひとつの指標となる.当院においては,大動脈基部径が正常値の200%(またはZ score 10)を越えた時点で,大動脈基部置換の適応検討を行い,大動脈弁・僧帽弁逆流の程度,使用することになる人工血管のサイズ(将来的に再度大動脈基部置換が必要になるかどうか?)などを総合して,症例ごとに手術適応を判断している.結果的には,小児期に大動脈基部置換を行ったほぼ全例で成人の大動脈基部置換の適応(大動脈解離の家族歴がある場合40~45 mm,大動脈解離の家族歴がない場合45~50 mm)を満たしていた.Loeys–Dietz症候群では,Marfan症候群に比べて大動脈解離のリスクが高いため成人体格における40 mmを目安に手術適応を考えている.
内科的治療で管理の難しい重度の僧帽弁閉鎖不全症は手術適応と判断する.第一選択は僧帽弁形成術で多くの症例で有効だが,形成術が有効でなかった場合には,術中判断で弁置換が必要となる可能性がある.そのような際に機械弁と生体弁のどちらを使用するのかについて,体格・性別・両親の希望などを踏まえてあらかじめ計画しておくことが重要である.
当院では,生涯にわたるフォローを見据えて,15~18歳で小児科から循環器内科への転科を行っている.この時期は,保護者から離れるなどフォローが途切れがちな時期なので,病名・病態の理解,定期フォローアップの必要性,突然死の可能性,将来的に外科手術が必要になる可能性,遺伝子検査の結果・児への遺伝リスクなどについて,本人が十分に理解していることを確かめることが重要である.疾患理解や自立度の確認など,可能であれば移行期支援外来のサポートも受けることが望ましい.また,特に遺伝については,移行期の患者が理解するのが難しいポイントでもあるために,当院では遺伝カウンセラーからも説明を行うようにしている.
Marfan症候群において,妊娠は大動脈解離のリスクとなることが知られており,妊娠前の大動脈基部径が40 mm未満で約1%,40 mm以上では約10%の頻度で大動脈解離を認めるとされる.そのため大動脈基部径が40 mm以上の患者では,妊娠前の基部置換を検討する.一方で,40 mm未満の症例でも大動脈解離のリスクはある点,大動脈基部置換を行ってもB型解離を起こす可能性は残っている点にも留意が必要である16).これらを含めた妊娠リスクに関して,思春期から繰り返し説明を行う必要がある.当院では,計画的妊娠の必要性,避妊の方法,妊娠前にアンギオテンシン受容体拮抗薬をβ遮断薬に切り替える必要がある点などについても併せて説明を行っている.
Marfan症候群は一生涯にわたって進行する多系統疾患で,年齢によって診るポイントが異なる.遺伝子検査の進歩により,Marfan類縁疾患との鑑別や遺伝子変異に基づく重症度分類などが可能になってきた.特に大動脈基部拡張の評価・治療が重要で,内科的・外科的治療の選択肢を踏まえながらフォローする必要がある.また,妊娠リスクに関する理解など,小児期から成人移行を見据えた管理が必要である.
本稿について,開示すべき利益相反はない.
1) Milewicz DM, Cecchi AC: Heritable Thoracic Aortic Disease Overview, in Adam MP, Feldman J, Mirzaa GM, et al (eds): GeneReviews(R)[Internet], Seattle (WA), University of Washington, Seattle, 1993–2024
2) Loeys BL, Dietz HC, Braverman AC, et al: The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010; 47: 476–485
3) Takeda N, Inuzuka R, Yagi H, et al: Clinical impact of copy number variation on the genetic diagnosis of syndromic Aortopathies. Circ Genom Precis Med 2021; 14: e003458
4) Schrijver I, Liu W, Odom R, et al: Premature termination mutations in FBN1: Distinct effects on differential allelic expression and on protein and clinical phenotypes. Am J Hum Genet 2002; 71: 223–237
5) Takeda N, Inuzuka R, Maemura S, et al: Impact of pathogenic FBN1 variant types on the progression of aortic disease in patients with Marfan syndrome. Circ Genom Precis Med 2018; 11: e002058
6) Taniguchi Y, Takeda N, Inuzuka R, et al: Impact of pathogenic FBN1 variant types on the development of severe scoliosis in patients with Marfan syndrome. J Med Genet 2023; 60: 74–80
7) Roman MJ, Devereux RB, Kramer-Fox R, et al: Two-dimensional echocardiographic aortic root dimensions in normal children and adults. Am J Cardiol 1989; 64: 507–512
8) Ogino H, Iida O, Akutsu K, et al: Japanese Circulation Society, the Japanese Society for Cardiovascular Surgery, the Japanese Association for Thoracic Surgery and the Japanese Society for Vascular Surgery Joint Working Group: JCS/JSCVS/JATS/JSVS 2020 Guideline on Diagnosis and Treatment of Aortic Aneurysm and Aortic Dissection. Circ J 2023; 87: 1410–1621
9) LoPresti MA, Ghali MZ, Srinivasan VM, et al: Neurovascular findings in children and young adults with Loeys-Dietz syndromes: Informing recommendations for screening. J Neurol Sci 2020; 409: 116633
10) Maron BJ, Chaitman BR, Ackerman MJ, et al: Working Groups of the American Heart Association Committee on Exercise, Cardiac Rehabilitation, and Prevention; Councils on Clinical Cardiology and Cardiovascular Disease in the Young: Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109: 2807–2816
11) Lacro RV, Dietz HC, Sleeper LA, et al: Pediatric Heart Network Investigators: Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med 2014; 371: 2061–2071
12) Brooke BS, Habashi JP, Judge DP, et al: Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med 2008; 358: 2787–2795
13) Ono M, Goerler H, Boethig D, et al: Current surgical management of ascending aortic aneurysm in children and young adults. Ann Thorac Surg 2009; 88: 1527–1533
14) Lange R, Badiu CC, Vogt M, et al: Valve-sparing root replacement in children with aortic root aneurysm: mid-term results. Eur J Cardiothorac Surg 2013; 43: 958–964
15) Patel ND, Arnaoutakis GJ, George TJ, et al: Valve-sparing aortic root replacement in children: Intermediate-term results. Interact Cardiovasc Thorac Surg 2011; 12: 415–419
16) Sayama S, Takeda N, Iriyama T, et al: Peripartum type B aortic dissection in patients with Marfan syndrome who underwent aortic root replacement: A case series study. BJOG 2018; 125: 487–493


This page was created on 2025-04-15T11:01:42.023+09:00
This page was last modified on 2025-05-23T16:20:48.000+09:00
このサイトは(株)国際文献社によって運用されています。