心筋症の遺伝子診断Genetic Testing for Cardiomyopathy
富山大学医学部小児科Department of Pediatrics, Faculty of Medicine, University of Toyama, Toyama, Japan
発行日:2024年5月31日Published: May 31, 2024
心筋症とは,心機能障害を伴う心筋疾患と定義され,原因はしばしば遺伝性である.小児の心筋症では様々な遺伝子に様々な変異があり,遺伝的多様性が特徴である.遺伝形式も常染色体顕性,常染色体潜性,X連鎖性,ミトコンドリア性と様々である.遺伝性心筋症の特徴として,同一遺伝子内の異なる変異は異なる病型を示すこと,遺伝子変異の多くが稀で同一のホットスポットや変異を有することは稀であること,同一の家族内でも様々な浸透率を示すことが挙げられる.また,同一の遺伝子変異を有していたとしても,臨床経過,転帰は同一家族内でも様々である.家族を含めた遺伝学的検査を行うことが重要であり,遺伝学的背景を念頭においた包括的な診療が不可欠である.
Cardiomyopathy is a myocardial disease that is accompanied by impaired heart function and often has a genetic cause. In pediatric cardiomyopathy, the presence of various gene variants highlights genetic diversity as a characteristic feature. Genetic forms can be autosomal dominant, autosomal recessive, X-linked, or mitochondrial, among others. Genetic cardiomyopathy is characterized by different clinical presentations of different variants within the same gene. Most gene variants are rare, and having the same hotspot or variant is uncommon. Even within the same family, these genetic variants can exhibit varying degrees of penetrance. Furthermore, even if individuals within the same family share the same genetic variant, their clinical courses and outcomes can differ significantly. For comprehensive patient care, consideration of the genetic background by genetic testing, including family members, is crucial.
Key words: gene; pathogenic variant; dilated cardiomyopathy; hypertrophic cardiomyopathy; left ventricular noncompaction
© 2024 特定非営利活動法人日本小児循環器学会© 2024 Japanese Society of Pediatric Cardiology and Cardiac Surgery
心筋症は重症心疾患であり,小児においても予後不良の転帰をもたらす.症候性心筋症を呈する小児の40%近くは,心臓移植を受けるか,診断後2年以内に死亡する.心臓移植を受けた心筋症の小児の割合は過去10年間減少しておらず,心筋症は依然として1歳以上の小児における移植の主要な原因である1).小児心筋症の発生率は,小児100,000人に1人程度であり,リンパ腫,ウィルムス腫瘍,神経芽細胞腫などの小児癌の発生率と同等である.また,日本小児循環器学会での報告では,小児では,拡張型心筋症,肥大型心筋症,左室心筋緻密化障害,拘束型心筋症の順に多い2).
2018年の日本循環器学会のガイドラインでは,心筋症は「心機能障害を伴う心筋疾患」と定義している3).(これは,家族歴や遺伝子変異の有無についての十分な検討と検索を重視し,二次性心筋疾患/二次性心筋症を鑑別したうえで,「原発性」心筋症を,肥大型心筋症,拡張型心筋症,不整脈原性右室心筋症,拘束型心筋症,の4つに分類している.また,小児の心筋症は,形態学的および臨床的に成人の心筋症に類似した症状を示すが,その転帰は大きく異なる.
遺伝子検査は,心筋症,不整脈障害,胸部大動脈瘤や胸部大動脈解離,家族性高コレステロール血症などのさまざまな遺伝性心血管疾患の臨床管理に有用であり,心筋症ガイドラインでも遺伝学的検査を推奨している4).しかし,循環器診療における遺伝学的検査の使用に関するエビデンスはまだまだ乏しい.
本稿では,遺伝子診断について最初に述べ,小児に多い拡張型心筋症,肥大型心筋症,左室心筋緻密化障害を取り上げる.
ヒトゲノムには体の各細胞に30億塩基対が含まれており,血縁関係のない人では約300万塩基対が異なる5).DNA変異はゲノム全体に発生する.ただし,少数のバリアントが,心筋症に寄与するバリアントの大部分を占める.疾患を引き起こす一塩基変異は集団内で非常に稀であることが多く,表現型に大きな影響を与える.機能喪失型バリアントには,(1)フレームシフトおよび下流の途中終止コドンを引き起こす挿入および欠失,(2)アミノ酸を途中終止コドンに変更するヌクレオチド置換,および(3)mRNAスプライシングを変更する標準的なスプライス部位バリアント,が含まれる.ミスセンス変異体は,コードされたタンパク質内でアミノ酸置換を引き起こし,その後の影響は最小限から大規模まで多岐にわたる.機能喪失型バリアントは多くの遺伝子の表現型をもたらす6).
染色体のペアにはゲノムDNAが含まれている.人間では,22対の染色体が常染色体を構成し,1つの染色体対(XおよびY)が性染色体を指す.さらに,ミトコンドリアには,酸化的リン酸化に関与するタンパク質のサブセットをコードする独特のゲノムが含まれている.常染色体顕性および常染色体潜性遺伝パターンは常染色体上の遺伝子に関係するが,X連鎖顕性およびX連鎖潜性遺伝はX染色体上の遺伝子を指す.ミトコンドリアは卵母細胞から母性遺伝するため,ミトコンドリアの遺伝は母親を通じて起こる.
ヒトのDNA配列(遺伝子型)と観察可能な形質(表現型)は関連している.ただし,同じ遺伝子変異が存在しても,個体間で表現型が異なる場合がある.可変表現性は,同じ遺伝子変異でみられる表現型の幅を表す.特定の遺伝子変異の浸透度と発現性に影響を与える要因はいくつかある.ゲノム内の二次変異,エピジェネティックな調節,および環境曝露はすべて,拡張型心筋症の表現型の不均一性に寄与する7).
タンパク質レベルでは,遺伝子の突然変異は,①アミノ酸配列に影響を及ぼさない,②アミノ酸配列を変化させるが機能的な影響を与えない,または③アミノ酸配列を変化させ深刻な機能異常を起こす,の結果のいずれかになる8).後者の場合,その影響は致死的であるか,疾患を引き起こす可能性がある.タンパク質の機能変化の重症度は,タンパク質内のアミノ酸の位置と,変異アミノ酸と野生型アミノ酸の物理的特性の違い(正電荷と負電荷,極性と非極性など)によって異なる.結果として生じる変異タンパク質は,軽度から重度までさまざまな機能の獲得または喪失を引き起こす可能性がある.心筋症の場合では,機能の喪失は,タンパク質の生成量が減少していること,またはタンパク質の活性が損なわれていることを意味する.機能獲得は,機能が変化し,変異タンパク質の異常な活性をもたらすことを示す.また,エネルギー生産と需要の変化,有毒な副産物の蓄積,エピジェネティックな要因など,時間の経過とともに病気の進行に寄与する非遺伝的要因もある9).
遺伝子診断のメリットとしては,病因の特定,リスク評価,親族の予測検査,家族計画に関するアドバイス等が挙げられる.
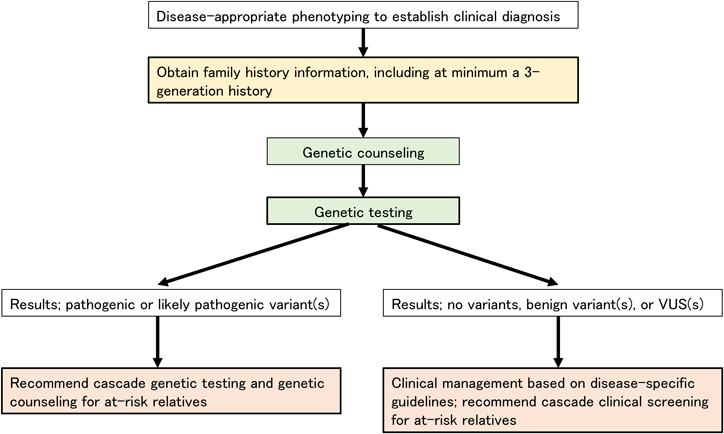
患者が遺伝性の心臓血管疾患を患っている可能性があるかどうかを評価するための重要な要素の1つは臨床症状の解析である.2番目の要素は少なくとも3世代にわたる包括的な家族歴の聴取である.また,患者は遺伝学的検査に同意する前に,遺伝学的検査の潜在的なリスクや限界を理解する必要がある.また,心筋症の臨床スクリーニングは,リスクのある一親等血縁者に対して推奨される.家族内に心筋症患者が2人以上見つかった場合,家族性の心筋症が想定される(Fig. 1)10).
遺伝情報の特徴として,①不変性:生涯変化しない,いつ検査をしても結果は変わらない,②予見性:将来の疾患などの発症を予測できる可能性がある,③共有性:家系で共通の情報を有する,検査を受ける当事者以外の血縁者にも影響を与えうる,がある.これらの特徴を考慮し検査の実施を慎重に判断する.染色体異常ではGバンド分染が,微小欠失症候群ではFISH法が,単一遺伝子疾患ではサンガーシーケンスや次世代シーケンスによる検査を考慮する.
2015年にアメリカ臨床遺伝学会はそれまで広く用いられていた多型,変異という表記をすべてバリアントに統合し,Pathogenic, Likely Pathogenic, Likely Benign, Benign, Unknown Significanceと5つのカテゴリに分類し表記を勧奨するという提言を公表した11).これはバリアントの集団中における頻度,コンピューター上のツールを用いたバリアントの有害度の予測結果,バリアントのタンパク質の機能に及ぼす影響に関する実験的な検証の有無,単一遺伝子疾患の家系における疾患との連鎖,新生バリアントであるか,同一遺伝子上の別のバリアントとのシス/トランスの関係,ほかの信頼できる情報源における評価などの項目ごとにランク付けを行い,それらの組み合わせによる上記の5つのカテゴリーに分類するものである.
単一遺伝子疾患の遺伝形式としては,常染色体顕性遺伝,常染色体潜性遺伝,X連鎖性顕性遺伝,X連鎖性潜性遺伝,Y連鎖遺伝,ミトコンドリア遺伝の形式が挙げられる.心筋症では,常染色体顕性遺伝,X連鎖性潜性遺伝がよくみられ,ここでは常染色体顕性遺伝,X連鎖性潜性遺伝の特徴について簡単に述べる.
常染色体顕性遺伝は,ヘテロ接合で発症し,遺伝子変異のある常染色体が一つでもあれば男女にかかわらず発症する.少なくとも両親のどちらかが発症し,どの世代にも罹患者が存在し,各罹患者は罹患した親を持つ.男女等しくいずれの性の子も受け継ぐ可能性があり,罹患した親のどの子もその形質を受け継ぐ可能性がある.また,罹患していない正常な親から子へは疾患を受け継がない.孤発例の大部分は新生突然変異による.
常染色体顕性遺伝疾患の表現型においては心筋症では以下の因子が重要である.
・浸透率:変異アレルを有する者のうち,疾患に罹患するものの割合.
・表現度:同一家系内でも症状の有無,あるいは症状の軽重に違いがあること.
・性腺モザイク:個体発生時に生殖細胞系列で突然変異が起こり,性腺において野生型アレルをもつ配偶子と変異アレルをもつ配偶子がモザイク状に混在すること.
・遺伝的異質性:臨床的に同じ疾患でありながら,異なる複数の責任遺伝子を有する場合を座位異質性という.臨床的には同じ疾患でも責任遺伝子内の変異を起こす塩基あるいは場所が異なる場合はアレル異質性という.
また,顕性遺伝疾患の発症メカニズムとしては,ハプロ不全と優性阻害効果,機能獲得型変異がある.
・ハプロ不全:2つのアレルの一方にナンセンス変異があり機能のないタンパク質が形成され,野生型アレルから作られるタンパク質の量では不足分を補填できずに機能不全を起こし表現型を示す.
・優性阻害効果:タンパク質が多量体を形成して機能する時などに,変異タンパク質の存在によって正常タンパク質の機能が阻害されてしまう現象.
・機能獲得型変異:タンパク質の正常な機能が亢進するような変異をいう.肥大型心筋症にみられる.
X連鎖性潜性遺伝は,ヘミ接合の男性で発症し,遺伝子変異のあるX染色体がY染色体と接合する.男性にのみ発症し,遺伝子変異のあるX染色体がX染色体と接合する女性は保因者となる.しかし,保因者女性のランダムなX染色体不活性化により,臨床症状には幅が生じることがある.発生率としては女性より男性が多い(患者はほとんどがXが一つだけの男性).女性は,通常は罹患しないが,時に男性より軽い症状を呈し発症することがある.通常は父から息子への直接伝達はなく,罹患した男性から娘すべてに伝達される.保因者女性を通じて何世代も伝達される.孤発例の場合は新生突然変異によるものが多い.
心筋症は病型のオーバーラップがしばしばみられる.例えば肥大型心筋症の患者は拘束型心筋症の表現を合わせ持つことがある.心筋症においては,病勢が進行すると他の病型に移行することがある.常染色体顕性遺伝の心筋症においては,たとえ同一の変異を有する家系であっても浸透率はさまざまである.
遺伝性心筋症の特徴として以下の三つが挙げられる.一つ目は,同一遺伝子内の異なる変異は異なる病型を示す.たとえば,サルコメア蛋白の一つである心臓トロポニンI(TNNI3)の遺伝子変異は,肥大型心筋症,拡張型心筋症,拘束型心筋症の原因となる12–14).二つ目は,各々の心筋症は各々の遺伝子変異により引き起こされる.遺伝子変異の多くが,稀で,特定の家族に限られ,同一のホットスポットや変異を有することは稀である.例えば,50以上の遺伝子が拡張型心筋症を引き起こし,また同一遺伝子内でもさまざまな遺伝子変異が拡張型心筋症を引き起こす.三つ目は,遺伝性心筋症は同一の家族内でもさまざまな浸透率を示す.同一の遺伝子変異を有していたとしても,臨床経過,転帰は同一家族内でも様々である.
肥大型心筋症は,著明な心筋肥大により心室内腔が狭小化し,拡張障害を来している疾患である.心室への流入血液量が減少し,心拍出量の低下を来す.小児期では無症状のことが多いが,運動中の失神や突然死が初発症状のことがある.罹患率は一般人口の0.2%と高頻度の報告がある15).小児期から若年成人期発症例では,乳児期に一つのピークがあり,それ以降は10歳から25歳で発症するものが多い.
小児肥大型心筋症の原因疾患は多様であり,特発性(74%)の他に,二次性として,代謝疾患(9%),症候群(9%),神経筋疾患(8%)が含まれる.乳幼児期診断例には,症候性(Noonan症候群,糖原病など)の肥大型心筋症類縁疾患が多く,学齢期診断例には,学校心臓検診で発見される無症状の特発性肥大型心筋症や,運動中の失神や突然死が初発症状である特発性肥大型心筋症が含まれる.最近の遺伝子-表現型連関の臨床的妥当性のエビデンスの強さに基づいた遺伝子のカテゴリー分類では,決定的な証拠および強力な証拠がある遺伝子として分類されたのは8遺伝子(MYBPC3, MYH7, TNNT2, TNNI3, TPM1, ACTC1, MYL2, MYL3)であり,3遺伝子(CSRP3, TNNC1, JPH2)は中程度の証拠が認められた(Table 1)16).心臓突然死イベントは5年間で9.1%発症し,診断時年齢,非持続性心室頻拍の記録,原因不明の失神,中隔径Zスコア,左心室後壁径Zスコア,左房径Zスコア,左心室流出路ピーク勾配,病的バリアントの存在が予測因子であり,左室流出路狭窄と心臓突然死の家族歴が成人とは異なっている17).初診時に肥大型心筋症の診断基準を満たさなかったサルコメア遺伝子変異保因者でも15年で半数が肥大型心筋症を発症するとされており,最初のスクリーニングにて陰性であっても,長期的にわたる定期的スクリーニングを考慮すべきである18).
| 原因遺伝子 | 染色体上の位置 | 遺伝形式 | コードする蛋白 | 心筋症病型 | ||
|---|---|---|---|---|---|---|
| ACTC1 | 15q14 | AD | actin, alpha, cardiac muscle 1 | HCM | DCM | LVNC |
| ACTN2 | 1q42-q43 | AD | actinin alpha 2 | DCM | ||
| ALPK3 | 15q25.3 | AR | alpha kinase 3 | LVNC | ||
| BAG3 | 10q25.2-q26.2 | AD | BCL2 associated athanogene 3 | DCM | ||
| CSRP3 | 11p15.1 | AD | cysteine and glycine rich protein 3 | HCM | ||
| DES | 2q35 | AD | desmin | LVNC | ||
| DMD | Xp21.2 | XR | dystrophin | DCM | LVNC | |
| DMPK | 19q13.32 | AD | DM1 protein kinase | LVNC | ||
| DSP | 6p24 | AR | desmoplakin | DCM | LVNC | |
| DTNA | 18q1 | AD | dystrobrevin alpha | LVNC | ||
| FLNC | 7q32.1 | AD | filamin C | DCM | ||
| HCN4 | 15q24.1 | AD | hyperpolarization activated cyclic nucleotide gated potassium channel 4 | LVNC | ||
| JPH2 | 20q13.12 | AD | junctophilin 2 | HCM | DCM | |
| LAMP2 | Xq24 | XD | Lysosomal-associated membrane protein 2 | LVNC | ||
| LDB3 | 10q22.3-q23.2 | AD | LIM domain binding 3 | LVNC | ||
| LMNA | 1q22 | AD | lamin A/C | DCM | LVNC | |
| MIB1 | 18q11.2 | AD | MIB E3 ubiquitin protein ligase 1 | LVNC | ||
| MYBPC3 | 11p11.2 | AD | myosin binding protein C, cardiac | HCM | LVNC | |
| MYH7 | 14q12 | AD | myosin heavy chain 7 | HCM | DCM | LVNC |
| MYL2 | 12q24.11 | AD | myosin light chain 2 | HCM | ||
| MYL3 | 3p21.3-p21.2 | AD | myosin light chain 3 | HCM | ||
| NEXN | 1p31.1 | AD | nexilin F-actin binding protein | DCM | ||
| NKX2-5 | 5q35.1 | AD | NK2 homeobox 5 | LVNC | ||
| NNT | 5p12 | AR | nicotinamide nucleotide transhydrogenase | LVNC | ||
| NONO | Xq13.1 | XL | non-POU domain containing octamer binding | LVNC | ||
| OBSCN | 1q42.13 | AD | obscurin, | LVNC | ||
| PKP2 | 12p11 | AD | plakophilin 2 | LVNC | ||
| PLEKHM2 | 1p36.21 | AD | pleckstrin homology and RUN domain containing M2 | LVNC | ||
| PLN | 6q22.1 | AD | phospholamban | DCM | LVNC | |
| PRDM16 | 1p36.32 | AD | PR/SET domain 16 | LVNC | ||
| RBM20 | 10q25.2 | AD | RNA binding motif protein 20 | DCM | LVNC | |
| RYR2 | 1q43 | AD | ryanodine receptor 2 | LVNC | ||
| SCN5A | 3p21 | AD | sodium voltage-gated channel alpha subunit 5 | DCM | LVNC | |
| TAZ | Xq28 | XR | tafazzin | LVNC | ||
| TBX20 | 7p14.2 | AD | T-box transcription factor 20 | LVNC | ||
| TBX5 | 12q24.21 | AD | T-box transcription factor 5 | LVNC | ||
| TMEM70 | 8q21.11 | AR | transmembrane protein 70 | LVNC | ||
| TNNC1 | 3p21.1 | AD | troponin C1, slow skeletal and cardiac type | HCM | DCM | |
| TNNI3 | 19q13.4 | AD, AR | troponin I3, cardiac type | HCM | DCM | |
| TNNT2 | 1q32 | AD | troponin T2, cardiac type | HCM | DCM | LVNC |
| TPM1 | 15q22.1 | AD | tropomyosin 1 (alpha) | HCM | DCM | LVNC |
| TTN | 2q31 | AD | titin | DCM | LVNC | |
| VCL | 10q22-q23 | AD | vinculin | DCM | ||
| AD, 常染色体顕性;AR, 常染色体潜性;XL, X連鎖性. | ||||||
β-ミオシン重鎖(MYH7;15~25%)と心臓ミオシン結合タンパク質C(MYBPC3;15~25%).さらに,心筋トロポミオシン(TPM1;~5%),心筋トロポニンT(TNNT2;~5%)およびトロポニンI(TNNI3;~5%),ミオシン調節軽鎖(RLC)およびミオシン必須軽鎖(ELC),ミオシン軽鎖(MYL2およびMYL3; 1~2%)の遺伝子が影響を受ける19).
MYBPC3の遺伝子変異は,肥大型心筋症では最もよく認められる遺伝子変異である.MYBPC3は,ミオシンを介した収縮の安定化や調節を行っていると考えられている.他の肥大型心筋症の遺伝子変異ではミスセンス変異によりアミノ酸が置換されるのとは対照的に,MYBPC3の多くはナンセンス変異かフレームシフトであり,不完全な蛋白が産生される20–22).近年,MYBPC3のバリアントは心室不整脈や失神が高率に認められ,心臓突然死のリスクが高いことが複数報告されている23, 24).
MYH7の遺伝子変異は肥大型心筋症全体の原因の約20%を占める.一般的に,MYH7遺伝子変異は,著明な肥大を伴う,比較的若年に発症する典型的な肥大型心筋症と関連がある25).主要心イベントの発生率も高いとの報告例もある26).また,成人でのメタ解析では心室頻拍が多いとの報告もある27).MYH7の遺伝子変異は肥大型心筋症だけに限らず,拡張型心筋症や心筋緻密化障害,ミオパチーにも見られる.
thin filamentのトロポニン複合蛋白をコードするTNNT2とTNNI3遺伝子も表現型の多様性があり,肥大型心筋症,拡張型心筋症,拘束型心筋症,心筋緻密化障害にも認められる12).他の多くのサルコメア蛋白のように,TNNT2変異はしばしば比較的軽症の拡張型心筋症を示すが心臓突然死のリスクは高い.トロポニンの遺伝子変異によりthin filament上のカルシウム結合能は肥大型心筋症の場合は増強し,拡張型心筋症の場合は減弱する28).
ミオフィラメントの遺伝子変異が陰性の肥大型心筋症家系の40%は,サルコメア周囲の遺伝子のZ帯やカルシウムを扱う蛋白をコードする遺伝子のCSRP3, TCAP, ANKRD1, MYOZ2などの変異で占められている29–32).
PRKAG2の変異は,グリコーゲンの蓄積や心筋細胞の錯綜配列の欠如により,常染色体顕性遺伝の形式で家族性の肥大型心筋症を引き起こす33).PRKAG2の遺伝子変異は心電図上,心室の早期興奮,徐脈傾向,進行性の伝導障害を伴ったWPW症候群も合併する.
Danon病は,X連鎖性の肥大型心筋症であり,ミオパチー,知的障害を伴う.LAMP2の遺伝子異常により,ライソゾームの膜レセプターを介したオートファジー機能異常が原因とされている.Danon病は,心室の早期興奮,不整脈,拡張型心筋症へと進行する著明な心筋肥大を特徴とし,通常の肥大型心筋症より予後不良であり,成人期早期にしばしば心移植が必要となる.
Fabry病の原因であるGLAの遺伝子異常は,全身性のX連鎖性のライソゾームの蓄積障害であり,心筋症,不整脈,腎障害,肢端触覚異常や被角血管腫,毛細血管拡張症といった皮膚症状,発汗低下,角膜混濁や脳血管障害を特徴とする.Fabry病はライソゾームのhydrolase α-galactosidase Aの欠損や欠乏により,glycosphingolipidが蓄積することが原因とされる.典型的には男性に見られるが,女性患者も見られる.GLA変異は3%の肥大型心筋症の家系に見出されている34).Fabry病は,心筋症が唯一の症状であることもある.酵素補充療法により治療が可能である.
拡張型心筋症は心筋収縮不全により心拡大を来し,心不全が進行する予後不良の疾患である.左心不全症状が主体で,乳児では哺乳不良や体重増加不良,年長児では運動時の易疲労性が認められる.小児期では,いずれの年齢にも認められる.原因疾患にかかわらず,5年生存率は50%と不良である35, 36).家族性,心筋炎後など原因はさまざまである.死亡原因は,ポンプ機能の低下による心不全か,不整脈による突然死である.
拡張型心筋症の約30~40%に家族性があり,小児期ではミトコンドリア遺伝や常染色体顕性遺伝形式をとるものが主体であり,X連鎖性や常染色体潜性遺伝形式は稀である.
家族性拡張型心筋症については,50以上の原因遺伝子が報告されており,その多くがイオンチャネル,サルコメア,Zディスク,核タンパク質,デスモソームをコードしている(Table 1)10, 37).孤発性の拡張型心筋症の場合は15~25%,家族性拡張型心筋症の場合は20~40%であり,TTN, LMNA, MYH7, TNNT2が主要な遺伝子である38–40).最近の遺伝子-表現型連関の臨床的妥当性のエビデンスの強さに基づいた遺伝子のカテゴリー分類では,51の遺伝子のうち,12遺伝子が決定的証拠(BAG3, DES, FLNC, LMNA, MYH7, PLN, RBM20, SCN5A, TNNC1, TNNT2, TTN)または強い証拠(DSP)を有するものとして分類され,7つの遺伝子(ACTC1, ACTN2, JPH2, NEXN, TNNI3, TPM1, VCL)が中程度の証拠と分類されている41).TTNの遺伝子変異が最もよく見出されており,家族性拡張型心筋症の25%,弧発性拡張型心筋症の18%で報告されている42).伝導障害を伴う拡張型心筋症の場合,LMNA遺伝子変異が1/3に認められる42).低年齢で拡張型心筋症と診断された患者,特に拡張型心筋症の家族歴がある患者は,遺伝的病因のリスクが高くなる.拡張型心筋症の成人患者では病的バリアントを保有する場合,心不全,致死性不整脈,脳梗塞を多く発生し予後が悪いことが報告されている43).
TTNは,体内で発現される最大のタンパク質(約35,000アミノ酸)をコードする.TTNタンパク質は,ZディスクからMラインまでサルコメアの半分に広がり,Zディスク結合領域,サルコメアのアクチンフィラメントと重なるIバンドドメイン,ミオシンフィラメントと重なるAバンドドメイン,およびMライン結合の4つのドメインで構成される.TTNのAバンド領域の切断型バリアントがすべての拡張型心筋症の約15~20%を占める42, 44).
ラミンA/Cは,LMNA遺伝子によってコードされるタンパク質である.その病的バリアントは,さまざまな種類の組織や器官系に影響を与えるラミノパチーを引き起し,心筋症は最も一般的な合併症である45).拡張型心筋症患者の約6%で,LMNA変異が観察される45).突然変異は,多くの場合,心房細動,洞結節または房室結節の機能不全,および心室不整脈と関連している37).
拡張型心筋症症例の約10%は,ミオシン結合プロテインCをコードするMYBPC3,ミオシン重鎖をコードするMYH7,トロポニンTをコードするTNNT2,およびトロポニンIをコードするTNNI3といったサルコメア遺伝子から見いだされる46).
RBM20は,心筋細胞と骨格筋に豊富に含まれるRNAスプライシング因子で,TTN,カルシウム/カルモジュリン依存性プロテインキナーゼIIデルタ(CAMK2D),およびリアノジン受容体2(RYR2)のスプライシングを制御する.RBM20に病的バリアントを持つ患者は,拡張型心筋症および心室不整脈のリスクが高く,死亡率も高い47–50).
DSPは,皮膚および心筋細胞で高度に発現されるデスモソームの成分であるデスモプラキンをコードする.DSPの病的バリアントは当初,常染色体顕性の不整脈原性右室異形成患者で報告されたが,拡張型心筋症でも見いだされる.心室不整脈は約半数にみられる51).
FLNCは,心筋細胞および骨格筋で高度に発現され,膜タンパク質とサルコメアを結び付けるアクチン結合中間フィラメントをコードする.FLNCのミスセンスバリアントは,これまでミオパチーや肥大型心筋症と関連付けられてきたが,最近の研究ではFLNC切断型バリアントが心室不整脈を有する拡張型心筋症の重要な要因であると報告されている52–54).
ナトリウムチャネルアルファユニットをコードするSCN5Aの変異は,不整脈のリスク増加と関連している.病的バリアントはQT延長やブルガダ症候群などの症候群に関連しているが,一部の変異は家族性の拡張型心筋症を引き起こす37).
ジストロフィン遺伝子はDuchenneやBecker muscular dystrophyの責任遺伝子でもある.骨格筋疾患の多くを占め,いずれも小児期あるいは若年成人で発症するが,ほとんどの症例は25歳までに拡張型心筋症を発症する.ジストロフィンは細胞骨格蛋白であり,格子状のネットワークを形成して,筋細胞を支持し,サルコメアと筋細胞膜や細胞外基質との結合と収縮力の伝達において,最も重要な役割を果たしている.また,NO合成酵素と作用して,細胞シグナル伝達にも関与している55).ジストロフィンのN末端はサルコメアのアクチンと結合し,C末端はジストログリカン,サルコグリカン,シントロピン,ジストロブレビンなどのジストロフィン関連蛋白と結合している.さらに,これらの蛋白は,α-ジストログリカンを通じて,ラミニン(laminin)や細胞外基質と結びついている.これらジストロフィン関連蛋白の異常は,ジストロフィン同様に骨格筋疾患や心筋症の原因となる.
左室心筋緻密化障害は,心室壁の過剰な網目状の肉柱形成と深い間隙を形態的特徴とする心筋症である56).臨床像は無症状の症例から高度の心機能障害を有し心移植の対象になっている症例まであり,きわめて多彩である57, 58).発症率は,新生児では10万人に76人,乳児では0.81人,小児では0.12人,成人では14人と報告されている.小児では心筋症全体の9.5%を占めると考えられている.
弧発性と家族性と双方みられ,4割程度に家族例が認められる.X連鎖性の他,常染色体顕性,常染色体潜性あるいはミトコンドリア遺伝子異常が報告されており,遺伝的多様性が特徴である.近年の報告では,小児では4割程度に遺伝子変異異常があると考えられている59).内訳としては,サルコメア遺伝子異常(MYH7, MYBPC3, ACTC1, TTN, TPM1, TNNT2等)が半数を占め,1割が不整脈関連の遺伝子異常(HCN4, RYR2, SCN5A等),それ以外の心筋症関連遺伝子(DTNA, FKTN, LMNA, PRDM16等)が続き,その他,TAZ等のミトコンドリア病関連遺伝子も見みられる(Table 1).最近の遺伝子と心筋緻密化障害の関連性の妥当性を評価したカテゴリー分類では,11遺伝子(TAZ, MIB1, MYH7, RYR2, TTN, MYBPC3, TPM1, DES, DSP, NONO, ACTC1)が確定的,21遺伝子が中程度,140遺伝子が限定的と分類されている60).染色体異常やミトコンドリア病などの疾患に合併することがある.
左室心筋緻密化障害の原因であることが判明した最初の病的バリアントは,Xq28座位にあるX連鎖潜性遺伝を持つTAZ遺伝子であった.TAZはタンパク質タファジンをコードし,タファジンは主に心筋および骨格筋で発現し,カルジオリピンの代謝に関与する.Barth症候群は心筋症,成長遅延,好中球減少症,3-メチルグルタコン酸尿症を特徴とする61).また,Duchenne型やBecker型筋ジストロフィーを引き起こすDMDの遺伝子異常も男児において左室心筋緻密化障害を引き起こす.
左室心筋緻密化障害患者の家族で,拡張型心筋症または肥大型心筋症が診断されることがよくある.左室心筋緻密化障害と診断された小児の25%が左室心筋緻密化障害または別の心筋症の家族歴を有する.
遺伝学的検査が診療や治療方針決定に有用であると報告例は少ないが,近年散見されるようになっている.肥大型心筋症,拡張型心筋症および心筋緻密化障害において,バリアントが複数見いだされる症例では,予後が不良であるとの報告がある26, 62, 63).肥大型心筋症患者でMYBPC3のバリアントを伴う症例では心室不整脈や心臓突然死のリスクが高い23, 24).拡張型心筋症ではTTNのバリアントを有する小児の症例は成人とは対照的に予後が良くないとされている64).このように,心筋症の遺伝的素因を明らかにするために遺伝学的検査を行うことは,診療や予後予測,治療方針の決定に有用である.
心筋症の浸透率について近年検証がなされてきている.Lorenziniらは,肥大型心筋症を発症していないサルコメア遺伝子変異保因者において15年間の追跡でサルコメア遺伝子変異保因者の約50%が肥大型心筋症を発症することを明らかにした.遺伝子別では,MYBPC3 43.2%,MYH7 24.2%,TNNI3 13.7%,TNNT2 11.9%,TPM1 3.2%,MYL2 2.1%,ACTC1 0.4%,多重変異1.4%であった.男性と心電図異常が肥大型心筋症発症の高いリスクと関連していた.Shahらは,UK Biobankにおいて,一般成人集団における拡張型心筋症関連推定病原性遺伝子変異体の頻度と浸透率を検証し,拡張型心筋症遺伝子の推定病因バリアントを有する成人の6人中1人以上が,拡張型心筋症遺伝子型と関連する可能性のある早期拡張型心筋症の特徴を示した65).これらのことから,最初のスクリーニングで陰性であっても,定期的な心臓MRIを考慮すべきであるとしている.
心筋症の成因については,多遺伝子の影響について近年検証がなされてきている.拡張型心筋症の遺伝的不均一性を考慮すると,マイナー対立遺伝子頻度が1%以上の一般的なバリアントが拡張型心筋症表現型に部分的に寄与すると予測するのは合理的であると近年考えられるようになってきている.ゲノムワイド関連研究(GWAS)により,集団レベルで特発性拡張型心筋症に関連するいくつかの一般的な一ヌクレオチド変異体が同定されている66–68).多遺伝子リスクスコア(PRS)は,その疾患に関連する個人の一般的な変異の数に基づいて,疾患を発症する相対リスクを計算する10).現在,拡張型心筋症に関連する一般的な亜種の数は比較的少ないため,多遺伝子リスクスコアの適用は制限されているが,追加のゲノムワイド関連研究が完成するにつれて,この適用により拡張型心筋症のリスク予測が強化される可能性がある.Tadrosらは,肥大型心筋症1,733例,拡張型心筋症5,521例,および健常なUK Biobank参加者19,260例において,ゲノムワイド関連研究および複数形質解析を行った69).その結果,肥大型心筋症と関連する16の遺伝子座,拡張型心筋症と関連する13の遺伝子座,左室形質と関連する23の遺伝子座が同定された.Harperらは,2,780例の症例と47,486例の対照例を対象としたゲノムワイド関連研究により,肥大型心筋症に対する12のゲノムワイドで有意な感受性遺伝子座を同定した70).一塩基多型の遺伝率は,特にサルコメア陰性の肥大型心筋症において,強い多遺伝子性の影響を示した.多遺伝的リスクスコアは肥大型心筋症の発症率と相関していた.このように多遺伝子リスクスコアは,心筋症の表現型と相関することが示され,多遺伝子の影響が心筋症でも示唆されている.
近年,心筋症において,遺伝学的要因が徐々に明らかにされてきているが,遺伝子・表現型相関が明らかであるのは一部に過ぎず,診療の場においては有用なエビデンスは多くない.また,診療において,遺伝学的検査が行われているのは少数の症例に限られる.今後の心筋症の研究と診療の発展のために,丁寧な家系情報の集積および正確な臨床情報収集が不可欠であり,親族を含めた包括的な医療を心がけていくことが重要である.また,エビデンスがまだ確立されていないが診療や治療方針決定に役立つ報告もあり,データの蓄積により確固たるエビデンスが構築されることを期待する.
本稿について,開示すべき利益相反はありません.
1) Lipshultz SE, Law YM, Asante-Korang A, et al: Cardiomyopathy in children: Classification and diagnosis: A scientific statement from the american heart association. Circulation 2019; 140: e9–e68
2) 日本循環器学会/日本小児循環器学会合同ガイドライン:学校心臓検診ガイドライン(2016年版)循環器病の診断と治療に関するガイドライン.2016
3) 日本循環器学会/日本心不全学会合同ガイドライン:心筋症診療ガイドライン(2018年改訂版)循環器病の診断と治療に関するガイドライン.2019
4) Musunuru K, Hershberger RE, Day SM, et al: American Heart Association Council on Genomic and Precision Medicine; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular and Stroke Nursing; and Council on Clinical Cardiology: Genetic testing for inherited cardiovascular diseases: A scientific statement from the american heart association. Circ Genom Precis Med 2020; 13(4): e000067
5) Bainbridge MN, Davis EE, Choi WY, et al: Loss of function mutations in NNT are associated with left ventricular noncompaction. Circ Cardiovasc Genet 2015; 8: 544–552
6) Walsh R, Thomson KL, Ware JS, et al: Exome Aggregation Consortium: Reassessment of mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med 2017; 19: 192–203
7) Hershberger RE, Hedges DJ, Morales A: Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat Rev Cardiol 2013; 10: 531–547
8) Burghardt TP, Neff KL, Wieben ED, et al: Myosin individualized: Single nucleotide polymorphisms in energy transduction. BMC Genomics 2010; 11: 172. doi:10.1186/1471-2164-11-172
9) Chou C, Chin MT: Pathogenic mechanisms of hypertrophic cardiomyopathy beyond sarcomere dysfunction. Int J Mol Sci 2021; 22: 22
10) Aragam KG, Natarajan P: Polygenic scores to assess atherosclerotic cardiovascular disease risk: Clinical perspectives and basic implications. Circ Res 2020; 126: 1159–1177
11) Richards S, Aziz N, Bale S, et al: ACMG Laboratory Quality Assurance Committee: Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424
12) Kimura A, Harada H, Park JE, et al: Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet 1997; 16: 379–382
13) Mogensen J, Kubo T, Duque M, et al: Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Invest 2003; 111: 209–216
14) Murphy RT, Mogensen J, Shaw A, et al: Novel mutation in cardiac troponin I in recessive idiopathic dilated cardiomyopathy. Lancet 2004; 363: 371–372
15) Maron BJ: Hypertrophic cardiomyopathy. Lancet 1997; 350: 127–133
16) Ingles J, Goldstein J, Thaxton C, et al: Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genom Precis Med 2019; 12(2): e002460
17) Miron A, Lafreniere-Roula M, Steve Fan CP, et al: A validated model for sudden cardiac death risk prediction in pediatric hypertrophic cardiomyopathy. Circulation 2020; 142: 217–229
18) Lorenzini M, Norrish G, Field E, et al: Penetrance of hypertrophic cardiomyopathy in sarcomere protein mutation carriers. J Am Coll Cardiol 2020; 76: 550–559
19) Maron BJ, Maron MS: Hypertrophic cardiomyopathy. Lancet 2013; 381: 242–255
20) Marston S, Copeland O, Jacques A, et al: Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res 2009; 105: 219–222
21) Niimura H, Bachinski LL, Sangwatanaroj S, et al: Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med 1998; 338: 1248–1257
22) Charron P, Dubourg O, Desnos M, et al: Clinical features and prognostic implications of familial hypertrophic cardiomyopathy related to the cardiac myosin-binding protein C gene. Circulation 1998; 97: 2230–2236
23) Chida A, Inai K, Sato H, et al: Prognostic predictive value of gene mutations in Japanese patients with hypertrophic cardiomyopathy. Heart Vessels 2017; 32: 700–707
24) Field E, Norrish G, Acquaah V, et al: Cardiac myosin binding protein-C variants in paediatric-onset hypertrophic cardiomyopathy: Natural history and clinical outcomes. J Med Genet 2022; 59: 768–775
25) Watkins H, Rosenzweig A, Hwang DS, et al: Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med 1992; 326: 1108–1114
26) Mathew J, Zahavich L, Lafreniere-Roula M, et al: Utility of genetics for risk stratification in pediatric hypertrophic cardiomyopathy. Clin Genet 2018; 93: 310–319
27) Sedaghat-Hamedani F, Kayvanpour E, Tugrul OF, et al: Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: A meta-analysis on 7675 individuals. Clin Res Cardiol 2018; 107: 30–41
28) Robinson P, Griffiths PJ, Watkins H, et al: Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res 2007; 101: 1266–1273
29) Geier C, Gehmlich K, Ehler E, et al: Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum Mol Genet 2008; 17: 2753–2765
30) Hayashi T, Arimura T, Itoh-Satoh M, et al: Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J Am Coll Cardiol 2004; 44: 2192–2201
31) Arimura T, Bos JM, Sato A, et al: Cardiac ankyrin repeat protein gene (ANKRD1) mutations in hypertrophic cardiomyopathy. J Am Coll Cardiol 2009; 54: 334–342
32) Osio A, Tan L, Chen SN, et al: Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circ Res 2007; 100: 766–768
33) Blair E, Redwood C, Ashrafian H, et al: Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: Evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet 2001; 10: 1215–1220
34) Havndrup O, Christiansen M, Stoevring B, et al: Fabry disease mimicking hypertrophic cardiomyopathy: Genetic screening needed for establishing the diagnosis in women. Eur J Heart Fail 2010; 12: 535–540
35) Matitiau A, Perez-Atayde A, Sanders SP, et al: Infantile dilated cardiomyopathy: Relation of outcome to left ventricular mechanics, hemodynamics, and histology at the time of presentation. Circulation 1994; 90: 1310–1318
36) Lipshultz SE, Sleeper LA, Towbin JA, et al: The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med 2003; 348: 1647–1655
37) McNally EM, Mestroni L: Dilated cardiomyopathy: Genetic determinants and mechanisms. Circ Res 2017; 121: 731–748
38) Pugh TJ, Kelly MA, Gowrisankar S, et al: The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med 2014; 16: 601–608
39) Morales A, Kinnamon DD, Jordan E, et al: Variant interpretation for dilated cardiomyopathy: Refinement of the American College of Medical Genetics and Genomics/ClinGen Guidelines for the DCM Precision Medicine Study. Circ Genom Precis Med 2020; 13(2): e002480
40) Lek M, Karczewski KJ, Minikel EV, et al: Exome Aggregation Consortium: Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536: 285–291
41) Jordan E, Peterson L, Ai T, et al: Evidence-based assessment of genes in dilated cardiomyopathy. Circulation 2021; 144: 7–19
42) Herman DS, Lam L, Taylor MR, et al: Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012; 366: 619–628
43) Escobar-Lopez L, Ochoa JP, Mirelis JG, et al: Association of genetic variants with outcomes in patients with nonischemic dilated cardiomyopathy. J Am Coll Cardiol 2021; 78: 1682–1699
44) Haggerty CM, Damrauer SM, Levin MG, et al: Genomics-first evaluation of heart disease associated with titin-truncating variants. Circulation 2019; 140: 42–54
45) Lu JT, Muchir A, Nagy PL, et al: LMNA cardiomyopathy: Cell biology and genetics meet clinical medicine. Dis Model Mech 2011; 4: 562–568
46) Kayvanpour E, Sedaghat-Hamedani F, Amr A, et al: Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clin Res Cardiol 2017; 106: 127–139
47) Brauch KM, Karst ML, Herron KJ, et al: Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol 2009; 54: 930–941
48) Guo W, Schafer S, Greaser ML, et al: RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med 2012; 18: 766–773
49) Refaat MM, Lubitz SA, Makino S, et al: Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm 2012; 9: 390–396
50) Maatz H, Jens M, Liss M, et al: RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J Clin Invest 2014; 124: 3419–3430
51) Smith ED, Lakdawala NK, Papoutsidakis N, et al: Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation 2020; 141: 1872–1884
52) Ortiz-Genga MF, Cuenca S, Dal Ferro M, et al: Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol 2016; 68: 2440–2451
53) Begay RL, Tharp CA, Martin A, et al: FLNC gene splice mutations cause dilated cardiomyopathy. JACC Basic Transl Sci 2016; 1: 344–359
54) Golbus JR, Puckelwartz MJ, Dellefave-Castillo L, et al: Targeted analysis of whole genome sequence data to diagnose genetic cardiomyopathy. Circ Cardiovasc Genet 2014; 7: 751–759
55) Chang WJ, Iannaccone ST, Lau KS, et al: Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc Natl Acad Sci USA 1996; 93: 9142–9147
56) Ichida F, Hamamichi Y, Miyawaki T, et al: Clinical features of isolated noncompaction of the ventricular myocardium: Long-term clinical course, hemodynamic properties, and genetic background. J Am Coll Cardiol 1999; 34: 233–240
57) Chin TK, Perloff JK, Williams RG, et al: Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation 1990; 82: 507–513
58) Pignatelli RH, McMahon CJ, Dreyer WJ, et al: Clinical characterization of left ventricular noncompaction in children: A relatively common form of cardiomyopathy. Circulation 2003; 108: 2672–2678
59) Hirono K, Hata Y, Miyao N, et al: LVNC study collaborates*: Increased burden of ion channel gene variants is related to distinct phenotypes in pediatric patients with left ventricular noncompaction. Circ Genom Precis Med 2020; 13(4): e002940
60) Rojanasopondist P, Nesheiwat L, Piombo S, et al: Genetic basis of left ventricular noncompaction. Circ Genom Precis Med 2022; 15(3): e003517
61) Clarke SL, Bowron A, Gonzalez IL, et al: Barth syndrome. Orphanet J Rare Dis 2013; 8: 23
62) Burstein DS, Gaynor JW, Griffis H, et al: Genetic variant burden and adverse outcomes in pediatric cardiomyopathy. Pediatr Res 2021; 89: 1470–1476
63) Wang C, Hata Y, Hirono K, et al: for LVNC Study Collaborators: A wide and specific spectrum of genetic variants and genotype-phenotype correlations revealed by next-generation sequencing in patients with left ventricular noncompaction. J Am Heart Assoc 2017; 6: 6
64) Khan RS, Pahl E, Dellefave-Castillo L, et al: Genotype and cardiac outcomes in pediatric dilated cardiomyopathy. J Am Heart Assoc 2022; 11: e022854
65) Shah RA, Asatryan B, Sharaf Dabbagh G, et al: Genotype-First Approach Investigators: Frequency, penetrance, and variable expressivity of dilated cardiomyopathy: Associated putative pathogenic gene variants in UK Biobank participants. Circulation 2022; 146: 110–124
66) Villard E, Perret C, Gary F, et al: Cardiogenics Consortium: A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur Heart J 2011; 32: 1065–1076
67) Meder B, Ruhle F, Weis T, et al: A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur Heart J 2014; 35: 1069–1077
68) Xu H, Dorn GW 2nd, Shetty A, et al: A genome-wide association study of idiopathic dilated cardiomyopathy in African Americans. J Pers Med 2018; 8: 8
69) Tadros R, Francis C, Xu X, et al: Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat Genet 2021; 53: 128–134
70) Harper AR, Goel A, Grace C, et al: HCMR Investigators: Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat Genet 2021; 53: 135–142


This page was created on 2024-07-09T17:55:56.109+09:00
This page was last modified on 2024-09-09T18:41:06.000+09:00
このサイトは(株)国際文献社によって運用されています。