先天性心疾患を合併する染色体異常Chromosomal Abnormalities Associated with Congenital Heart Disease
東京都立小児総合医療センター 循環器科Department of Cardiology, Tokyo Metropolitan Children’s Medical Center ◇ Tokyo, Japan
発行日:2024年2月29日Published: February 29, 2024
先天性心疾患は,出生1,000人につき5~10人の頻度で発生し,生命予後に大きな影響を及ぼす先天異常である.先天性心疾患の多くは多因子遺伝により発症するが,一部は染色体異常または先天異常症候群の合併症として生じることから,特定の染色体上に座位する遺伝子の異常が,先天性心疾患の原因となることが示唆される.近年の分子遺伝学的研究の進歩により,染色体異常を端緒として,心血管疾患の候補遺伝子の同定や,心血管発生の分子メカニズムの解明が進められている.本稿では,染色体異常のうち,小児循環器医が日常診療で関与する頻度が高く,保険診療内で診断可能な疾患を取り上げ,それらの遺伝学的要因と先天性心疾患の特徴について述べる.
Congenital heart disease occurs in 5 to 10 out of every 1000 live births and has a significant impact on mortality risk throughout life. Although most cases are due to multifactorial inheritance, some appear to be caused by a chromosomal abnormality or congenital anomaly syndrome, implying that specific genes within impaired chromosomes are to blame. Recent advances in molecular genetics have allowed for the identification of genes in critical genomic regions of chromosomal abnormality that may be at the root of congenital heart diseases, as well as the clarification of molecular pathways in cardiovascular development. The current article examines the clinical characteristics and genetics of chromosomal abnormalities associated with congenital heart diseases that are frequently detected by pediatric cardiologists during genetic examinations covered by National Health Insurance.
Key words: chromosomal abnormality; congenital anomaly syndrome; congenital heart disease; trisomy; microdeletion
© 2024 特定非営利活動法人日本小児循環器学会© 2024 Japanese Society of Pediatric Cardiology and Cardiac Surgery
先天性心疾患は出生1,000人につき,5~10人の頻度で発生する.2021年に日本小児循環器学会で行われた小児科発生心疾患実態調査では,出生数811,640人に対して,先天性心疾患は9,580人で,発生率は1.39%であった1).先天性心疾患の60%は多因子遺伝であり,複数の遺伝子や遺伝子外の環境要因が関与して発生するが,他の要因として,遺伝子コピー数異常が15%,染色体異常が13%,単一遺伝子異常が12%と報告されている2).単独で先天性心疾患の原因となる多くの遺伝子は,胎生期の心臓発生に関与する転写因子やシグナル伝達分子,構造蛋白をコードし,心臓の原基における発現部位や時期が,多数の分子経路によって制御されている.これらの遺伝子異常が種々の先天性心疾患の原因になりうることは容易に想像される.しかし,心外合併症のない先天性心疾患における単一遺伝子異常の診断は研究室レベルにとどまり,現時点では診療への寄与は高くない3).一方で,先天性心疾患は,染色体異常や先天異常症候群に合併することが多く,その20%は心外合併症を伴う症候群性であると報告されている4).Table 1に先天性心疾患を合併する頻度が高い染色体異常を示した.近年の遺伝学的研究の進歩により,染色体異常,先天性異常症候群の責任領域・疾患遺伝子が明らかにされ,先天性心疾患発症の分子メカニズムの解明が進められている.
| Chromosomal abnormality (Congenital anomaly syndrome) | CHD | Frequency | Extracardiac features |
|---|---|---|---|
| Trisomy 21 (Down syndrome) | VSD, AVSD, PDA, ASD, TOF | 40–50% | Characteristic facial features, mental retardation, hypotonia, leukemia, hyperuricemia, atlantoaxial subluxation, hypothyroidism |
| Trisomy 18 (Edward syndrome) | VSD, PDA, TOF, DORV, CoA, BAV, BPV, polyvalvular disease | 80–90% | Characteristic facial features, mental retardation, growth disturbance, prominent occiput, overlapping fingers, rocker-bottom feet, central apnea, hepatoblastoma |
| Trisomy 13 (Patau syndrome) | VSD, ASD, PDA, HLHS | 80% | Characteristic facial features, growth disturbance, mental retardation, polydactyly, skin defects of posterior scalp, cleft lip, umbilical hernia, pancreatitis |
| Monosomy X (Turner syndrome) | CoA, BAV, AS, HLHS, aortopathy | 23–35% | Short stature, gonadal dysplasia, webbed neck, cubitus valgus, lymphedema |
| 1p36 deletion (1p36 deletion syndrome) | PDA, VSD, ASD, Ebstein disease, DCM, LVNC | 44% | Characteristic facial features (deep-set eyes, pointed chin), growth disturbance, mental retardation, microcephaly, hypotonia, seizures, hearing loss |
| 3p25 deletion (3p deletion syndrome) | VSD, AVSD, TA | 33% | Ptosis, polydactyly, mental retardation |
| Trisomy 3q21-qter (3q duplication syndrome) | Variable cardiovascular disease | Characteristic facial features (broad nasal bridge), mental retardation | |
| Deletion 4p16.3 (4p deletion syndrome, Wolf Hirschhorn syndrome) | ASD, VSD, PDA | 50% | Characteristic facial features (Greek warrior helmet like nose, hypertelorism), microcephaly, mental retardation, seizures |
| Deletion 5p (5p deletion syndrome) | VSD, PDA, ASD | 30–60% | Characteristic facial features (round face, micrognathia), microcephaly, high-pitched cry in infancy, mental retardation |
| Deletion 7q11.23 (Williams syndrome) | SVAS, PPS | 75% | Elfin face, hoarseness, infantile hypercalcemia, dental abnormalities, hypertension, visuospatial recognition disorder |
| 7q11.23 duplication | PDA, ASD, aortopathy | Characteristic facial features (straight eyebrows, broad forehead), mental retardation (speech delay), hypotonia | |
| Trisomy 8 mosaicism | VSD, PDA, CoA, PS, TAPVC, TrA | 25% | Characteristic facial features (hypertelorism, broad nasal root, micrognathia), camptodactyly |
| Trisomy 9 mosaicism | PDA, VSD, TOF, DORV | 65% | Prenatal growth retardation, low-set ears, joint contractures, severe mental retardation |
| Deletion 9p (9p deletion syndrome) | VSD, PDA, PS | 35–50% | Characteristic facial features (trigonocephaly, hypoplastic supraorbital ridges), mental retardation |
| Duplication 10q24-qter (10q duplication syndrome) | AVSD, VSD | 70% | Characteristic facial features (ptosis, short palpebral fissures), camptodactyly, mental retardation |
| Deletion 11q (Jacobsen syndrome) | HLHS, CoA, AS, VSD | 60% | Characteristic facial features (hypertelorism, carp-shaped mouth), thrombocytopenia, platelet dysfunction, mental retardation |
| Tetrasomy 12p mosaicism (Pallister-Killian syndrome) | VSD, ASD | 40% | Characteristic facial features (sparse anterior scalp hair, long philtrum), abnormal skin pigmentation, diaphragmatic hernia, mental retardation |
| Deletion 17p11.2 (Smith-Magenis syndrome) | VSD, PS, TOF | 25% | Characteristic facial features (flat midface, prominent forehead), sleep disorders, self-destructive behavior |
| Trisomy / tetrasomy 22p ter-q11 (cat eye syndrome) | TAPVC, ASD, VSD | 50% | Coloboma of iris, anal atresia, renal abnormalities |
| 22q10-q11 and 11q23-qter duplication (Emanuel syndrome) | VSD, ASD, TOF, PDA, TrA, TA | 60% | Microcephaly, growth disturbance, mental retardation, hearing loss, cleft palate |
| Deletion 22q11.2 (22q11.2 deletion syndrome) | TOF, TrA, IAA-B, VSD, Aortic arch anomaly | 75% | Characteristic facial features (small mouth, tubular nose, hypoplastic wing of the nose), thymic hypoplasia, nasopharyngeal dysfunction, hypocalcemia, mental retardation |
| 22q11.2 duplication | TOF, HLHS, VSD, PS | 15% | Variable phenotype |
| Deletion 22q13 | PDA, VSD, ASD | Characteristic facial features (pointed chin, long eyelashes), dolichocephaly, dysplastic ears, mental retardation | |
| AS, aortic stenosis; ASD, atrial septal defect; AVSD, atrioventricular septal defect; BAV, bicuspid aortic valve; BPV, bicuspid pulmonary valve; CoA, coarctation of aorta; CHD, congenital heart disease; DCM, dilated cardiomyopathy; DORV, double outlet right ventricle; HCM, hypertrophic cardiomyopathy; HLHS, hypoplastic left heart syndrome; IAA-B, interruption of aortic arch type B; LVNC, left ventricular noncompaction; MS, mitral stenosis; PDA, patent ductus arteriosus; PS, pulmonary stenosis; PPS, peripheral pulmonary stenosis; TA, tricuspid atresia; TrA, truncus arteriosus; SVAS, supravalvular aortic stenosis; TOF, tetralogy of Fallot; VSD, ventricular septal defect. (Modified from Pierpont et al45)) | |||
本稿では,先天性心疾患を合併する頻度の高い染色体異常症候群のうち,染色体検査が保険収載され,日常の診療で診断が可能な疾患を取り上げ,遺伝学的要因,心疾患の特徴を中心に述べる.遺伝子は斜体,遺伝子産物である蛋白質は正体,ヒト遺伝子名はすべて大文字,マウス遺伝子名は先頭のみ大文字で表記した.
胎生期の染色体異常の頻度は高く,自然流産した胎児の50%に認められる.常染色体数的異常であるトリソミーも出生前に流産することが多い.16トリソミーは胎生致死であり,出生後に診断される機会はない.Down症候群(21トリソミー),18トリソミーも,出生に至る頻度は,各々22%,5%である.Down症候群,18トリソミー,13トリソミーの出生頻度が高い理由として,これらの染色体上の遺伝子数が少なく,過剰による生命への影響が比較的軽度であることが考えられている.性染色体数的異常であるTurner症候群(Xモノソミー)はほとんどが流産し,出生に至る割合は0.7%であるが,ほかの性染色体異常の流産はまれであることから,X染色体が2本あることが胎生期生存に重要であることが示唆される5).
染色体検査は保険収載され,G分染法またはFISH(fluorescence in situ hybridization)法が用いられることが多い.G分染法では,末梢血リンパ球に細胞分裂促進因子を添加して細胞培養し,コルヒチンを加えて細胞分裂中期で止め,Giemsa染色を行う.数日間細胞培養を行ったのち標本を作製するため,解析まで時間を要する.染色される一つのバンドがDNAの約5~6メガ塩基対(Mb),すなわち500~600万塩基対に相当する.1バンドには約50個の遺伝子が存在する.濃く染まるバンドは,アデニン–チミン対が豊富で,遺伝子密度が低い.FISH法は,末梢血リンパ球のうち,細胞周期の間期核の細胞に対して,特定の染色体上のDNAに対する蛍光標識プローブを結合させる解析法で,細胞を培養せず,そのまま(in situ)用いるため,G分染法より早く,数日以内に結果が得られる.また,G分染法で検出困難な数万塩基対レベルの変化を同定できるが,プローブが用意されている特定の染色体異常の検査に限られる6).
胎児約200人に1人,出生700~1,000人に1人の頻度であり,最も出生頻度が高い染色体異常である.発生頻度は,母体年齢が35~39歳で270人に1人,40~44歳で100人に1人,45歳以上で50人に1人であり,母体年齢とともに増加する5, 7).標準型21トリソミーが最も多く(90~95%),ほとんどは孤発例である.相同染色体の第1減数分裂で配偶子が形成される際に,染色体不分離が生じ,片親の21番染色体2本ともう一方の親の21番染色体1本の計3本が受け継がれる.約90%で過剰21番染色体は母由来であり,母体年齢の増加に伴って染色体不分離の頻度は増加する.
転座型21トリソミー(3~5%)は,21番染色体の長腕が,14番,あるいは21番染色体の長腕と,お互いに小さな短腕を失って結合した派生染色体(Robertson型転座)を生じ,正常な14番,21番,および14番と21番が結合した派生染色体3本をもつ卵子または精子が形成される.これらの配偶子が正常核型の配偶子と受精し,21番染色体長腕が過剰になり発症する.ほかに生じうる組み合わせの14トリソミー,14モノソミー,21モノソミーは胎内死亡し,出生しない.同様に13, 15, 22番染色体も小さな短腕を持つが,21番と派生染色体を形成することはまれである.孤発例が多いが,25%で片親が転座保因者であるとされている.モザイク型21トリソミー(1~2%)は,トリソミー細胞と正常細胞が混在している状態で,体細胞分裂時の染色体不分離によると考えられている7, 8).
出生前診断として,非侵襲的であるが非確定検査である胎児エコー検査,母体血清マーカー検査,非侵襲性出生前遺伝学的検査(non-invasive prenatal genetic test: NIPT),侵襲的であるが確定検査である絨毛検査,羊水検査がある.NIPTは母体血液中に混入する胎児DNA断片を用いる検査で,遺伝カウンセリング体制の整った認可施設では,21, 18, 13トリソミーの検査のみ行われている.トリソミー陽性と判定された場合でも,羊水検査で確定診断する必要がある.NIPTによるDown症候群の陽性的中率(検査陽性で,実際にDown症候群と診断される確率)は,妊娠週数,母体年齢により異なるが,国内調査では約97%,陰性的中率(検査陰性で,実際にDown症候群ではない確率)は99.99%以上であった9).
約40~45%に先天性心疾患を認め,欧米の報告ではそのうち房室中隔欠損(AVSD)の頻度が最も高く,約45%を占め,Down症候群のAVSDの75%以上は完全型である.また,AVSDの50%にDown症候群が合併するとされている10).Table 2にDown症候群における先天性心疾患の頻度を示す11–13).スウェーデンのDown症候群2588例を対象とした調査では,AVSD 582例(42.0%),心室中隔欠損(VSD)307例(22.1%),心房中隔欠損(ASD)224例(16.2%),動脈管開存(PDA)70例(5.0%),Fallot四徴(TOF)36例(2.6%)であった12).国内のDown症候群1028例を対象とした全国調査では,VSDの頻度が最も高く,378例(36.8%)で,以下,AVSD 305例(29.7%),PDA 108例(10.5%),ASD 106例(10.3%),TOF 76例(7.4%)であった13).Down症候群では完全大血管転位(TGA)はまれとされており,国内調査でも認められなかったが13),スウェーデンの調査では26例(1.0%)に認められており12),心疾患発症における人種差の関与も示唆される.また,国内調査では,約60%の症例で複数の心疾患が合併し,その大半は左右短絡疾患であった13).
| CHD | Bull11) | Bergström et al12) | Oana et al13) |
|---|---|---|---|
| AVSD | 45% | 42.0% | 29.8% |
| VSD | 35% | 22.1% | 37.0% |
| ASD | 8% | 16.2% | 10.4% |
| PDA | 7% | 5.0% | 10.6% |
| TOF | 4% | 2.6% | 7.4% |
| Aortic arch anomaly | 6.5% | frequent | |
| PH | 1.2–5.2% | 38% | |
| ASD, atrial septal defect; AVSD, atrioventricular septal defect; CHD, congenital heart disease; PDA, patent ductus arteriosus; PH, pulmonary hypertension; TOF, tetralogy of Fallot; VSD, ventricular septal defect. | |||
右鎖骨下動脈起始異常などの大動脈弓異常,左上大静脈遺残などの大静脈異常の合併も多く,大動脈弓異常の合併は6.5%と報告されている12, 13).右鎖骨下動脈起始異常は,胎児心エコーによる解析では,染色体異常のない胎児で1.02%であったのに対し,Down症候群胎児で23.6%と高頻度であった14).国内の2つの病院を対象とした調査では,Down症候群76例中23例(30%)に合併が認められた15).21番染色体の過剰が大動脈弓異常を来す機序は明らかにされていない.
Down症候群のAVSDでは,Rastelli分類A型に加えC型の頻度も高く,国内調査では,C型がA型よりやや頻度が高かった13).21番染色体上のDSCR1(RCAN1)やDYRK1A遺伝子が,トリソミーにより1.5倍発現することで,房室弁や心中隔形成に関与するCRELD1(3p25.3に座位)やNFATC1(18q23に座位)遺伝子の発現低下を来し,AVSDを発症する機序が推測されている16).また,Down症候群では,AVSDとTOFの合併が特徴的で,AVSDの約9~17%に認められた1, 12, 17).一方,AVSDとTOF合併例の74%はDown症候群であると報告されている18).胎児の前後軸決定を調節する転写因子HOXB1(17q21.32に座位)は右室流出路と房室中隔の発生に関与しており,21番染色体の過剰によりHOXB1の機能が低下し,TOFとAVSDを生じる可能性が推測されているが18),遺伝学的機序について今後の研究が待たれる.
Down症候群では,先天性心疾患に伴う肺高血圧の頻度が高く,その頻度は6~37.5%と報告されている17).本邦からの報告では,先天性心血管疾患を有するDown症候群患者の肺高血圧合併頻度は38%であった13).Down症候群に伴う肺高血圧の成因として,肺血管床の低形成や,血管作動性因子の不均衡による肺動脈攣縮が考えられている19, 20).Down症候群に伴うRastelli A型のAVSDでは肺高血圧を合併するリスクが高いとされている19).また,21番染色体上のCOL18A1, COL4A3, TIMP3, APPの各遺伝子がコードする血管新生抑制因子が肺組織で過剰発現し,肺血管床の発達が抑制されるという報告がある21).そのほか,Down症候群に合併する巨舌,中咽頭狭窄や,喉頭および気管軟化症,睡眠時無呼吸,先天的な肺胞低形成は,低酸素血症を来し,肺高血圧を増悪させる19).また,Down症候群のVSDでは,染色体異常のないVSDと比較して,肺血管コンプライアンス(1回右室拍出量/肺動脈脈圧),すなわち肺動脈の弾力性,伸展性が低く,術後も肺動脈圧が高く,在宅酸素療法導入が有意に多かったと報告されている22).
Down症候群では,左右短絡の持続により早期に肺血管閉塞性病変が進行すると考えられているが,VSD, AVSDを伴うDown症候群188例と非Down症候群94例の肺動脈組織所見を比較した研究では,肺血管閉塞性病変の進行は,Down症候群の有無ではなく,年齢,肺動脈圧に相関するということが示された23).また,VSD, AVSDに対して肺動脈絞扼術が行われたDown症候群23例を含む34例において,手術前後で肺動脈組織を解析した結果,生後6か月までに手術が行われた症例では,肺動脈中膜や内膜肥厚の改善が観察された24).Down症候群であっても,生後6か月までの肺血流制限が,術後の肺高血圧残存リスクを低減させることが示唆される.心内修復術後に留意すべき合併症に,肺動脈圧が発作性に上昇し,循環不全を呈する肺高血圧クライシスがあり,国内調査では,VSD術後における頻度は非Down症候群の0.2%に対して,1.1%と報告されている25).また,左心系のサイズが小さいアンバランス型AVSDは,Fontan型手術の候補であるが,肺高血圧のリスクが高いDown症候群では慎重に適応を決定する必要がある17).非Down症候群と比較すると,Down症候群ではFontan型手術後の死亡率が有意に高く(1.6% vs 12.3%),心不全,腎不全,感染症,乳び胸,長期の人工呼吸管理などの術後合併症が多いことが報告されている26).
胎児500人に1人,出生6,000~8,000人に1人の頻度で認められる27).出生時の染色体異常としてはDown症候群に続き,2番目に頻度が高い.出生児では女児が多く(女 : 男=3 : 1),男児より生存期間が長いとされているが,その理由は不明である28).標準型18トリソミーが最も多く(93.8%),減数分裂時の染色体不分離が原因で,約95%は母由来であり,母体年齢とともに頻度が増加する.モザイク型(4.6%)は,体細胞分裂時の染色体不分離で生じ,各臓器のモザイク細胞の比率により,表現型に幅がある.転座型の頻度は少ない(1.7%)8).18番染色体上の遺伝子の過剰と,先天性心疾患をはじめとする18トリソミーの合併症の発症機序は明らかにされていない.妊娠週数,年齢により異なるが,NIPTによる出生前診断の陽性的中率は,約88%,陰性的中率は99.99%以上と報告されている9).
約90%に先天性心疾患を合併する.VSD, ASD, PDAなどの肺血流増加型の単純型心疾患が多く,18トリソミー134例の国内調査では,VSDが75例(59%)を占め,そのうちの68%にASDやPDAを合併していた.また,複雑型心疾患では,両大血管右室起始(DORV)が11%に認められ,71%は肺血流増加型であり,21%に僧帽弁閉鎖(MA)を合併していた29).剖検においても,MAを合併したDORVを高頻度に認めたという報告がある30).また,房室弁尖の余剰組織,肥厚,粘液腫様変化,半月弁の二尖形態,弁尖余剰組織,肥厚などの弁形態異常を高頻度に認める(Fig. 1).国内調査では,房室弁や半月弁の形態異常を46%に認め,そのうち57%は複数の弁異常であった29).18トリソミー41例の剖検では,三尖弁38例(80%),肺動脈弁30例(73%),大動脈弁28例(68%),僧帽弁27(66%)に形態異常があり,38例(93%)で2つ以上の弁に,14例(34%)で4弁すべてに異常が認められた30).これらの多弁病変は,18トリソミーの68~100%に認められ31),特徴的な所見であるといえる.そのほか,頻度は少ないが,AVSD,三尖弁閉鎖,TOF,完全大血管転位(TGA)や左心低形成症候群(HLHS)も認められる28, 29).
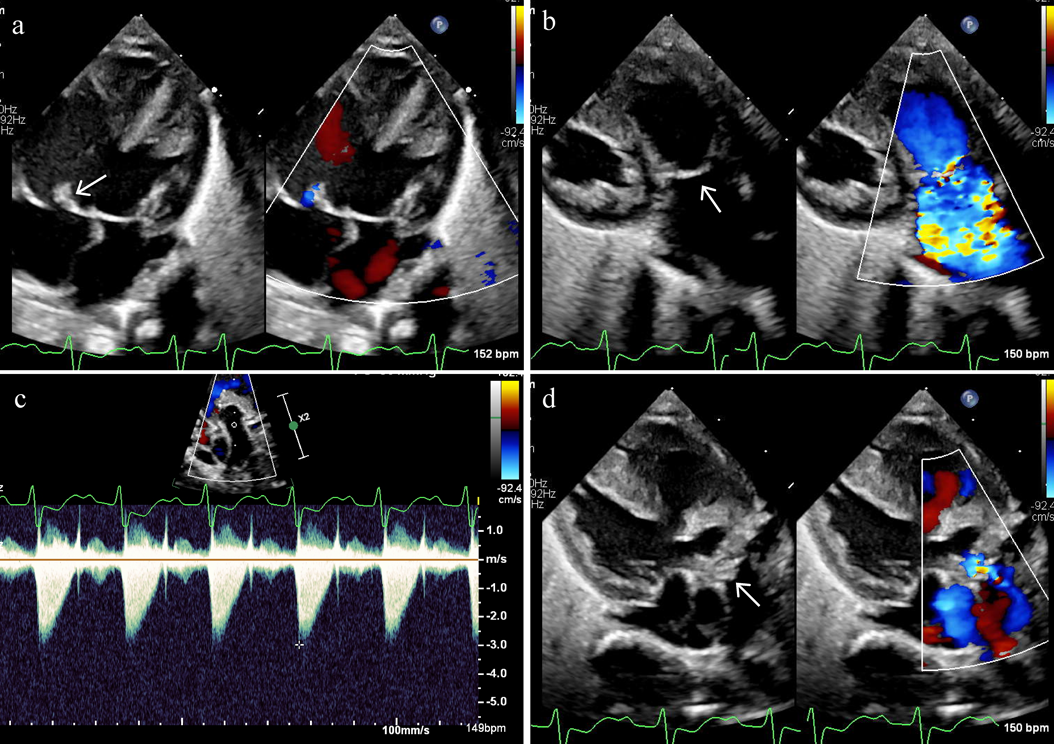
a) Hypertrophic septal leaflet of tricuspid valve (arrow). b) Thick and domed pulmonary valve (arrow). c) Continuous wave Doppler interrogation of pulmonary valve with peak velocity of 3.0 m/sec, suggesting moderate stenosis. d) Hypertrophic aortic valve (arrow).
肺血流増加型心疾患では,乳児期早期から肺高血圧の合併が多く,国内調査では52%に認められた29).剖検では,25例中8例(32%)に肺動脈中膜肥厚,内膜増生が認められ,その8例中6例は生後2か月未満の症例であった30).肺生検が行われた18トリソミー28例の肺動脈の組織学的検討では,13例(46.4%)に肺小動脈の低形成,4例(14.3%)に肺小動脈中膜形成不全(中膜平滑筋が欠如),15例(53.6%)に肺胞低形成,21例(75.0%)に肺胞壁肥厚を認めた.中膜形成不全の4例は全例死亡し,うち2例は心臓関連死であった32).中膜形成不全は,Down症候群94例の肺生検組織では認められず33),18トリソミーの一部に特徴的な所見であり,かつ予後不良の指標であることが示唆される.先天的に中膜平滑筋の発達が不良である肺小動脈では,高肺血流による物理的なストレスに対して,内膜の線維増生が招来され,肺血管閉塞性病変に移行する機序が推測される32, 34).一方で,肺高血圧を伴うVSDを合併した18トリソミー,Down症候群,染色体異常のない対照の3群の比較で,18トリソミーの肺体血流比,肺血管抵抗,肺血管コンプライアンスは,対照群と有意差がなく,Down症候群より有意に肺体血流比,肺血管コンプライアンスが高いことを示した報告がある35).肺動脈中膜形成不全は18トリソミーに特徴的な所見ではあるものの,14.3%にとどまったことから32),中膜異常を合併しない18トリソミーも多く,それらの症例ではDown症候群と肺動脈の反応性が異なること,手術により肺高血圧が改善する可能性が考えられた35).
18トリソミーの国内調査では18トリソミーで不整脈関連死が12%,突然死が15%認められた29).一方,米国の18トリソミー54例の検討では,明らかな不整脈関連死はなく,27%に頻脈性不整脈を認めたが,抗不整脈薬で再発なく治療可能であったと報告されている36).
近年は在宅医療を目指して姑息術や心内修復術が行う施設が増加している.米国の18トリソミー65例,13トリソミー30例の心臓手術後の調査では,心内修復術症例の術後生存率が姑息手術症例より有意に高いことが示され(15年生存率70.7% vs 30.8%),18トリソミーの術後遠隔期の生存期間(中央値)は16.2年であった37).本邦の報告では,18トリソミー,VSD 18症例に対して心内修復術を行い,術後生存期間(中央値)は,46.3か月であった38).心内修復術の有用性が示される一方,心臓手術のために入院した18トリソミーの死亡率は13%で,特に単純型心疾患の手術については,非トリソミーの場合より約10倍死亡率が高く,低年齢,平均肺動脈圧や肺血管抵抗値が高い場合,術前に人工呼吸管理を要した場合に死亡率が高いとされている38, 39).
出生児の中で,Down症候群,18トリソミーに次いで,3番目に頻度が高い常染色体異常で40),出生5,000~12,000人に1人の頻度で認められる41).標準型13トリソミーが最も多く(約80%),減数分裂時の染色体不分離が原因で,90%が母由来であり,母体年齢とともに頻度が増加する.他のトリソミーに比して転座型が多く(約5~20%),13番と14番のRobertson転座が主である.Down症候群や18トリソミーと異なり,モザイク型はまれである.13q遠位部部分トリソミー(13q14→qter),13q近位部部分トリソミー(13pter→q14)があり,生命予後は標準型よりよい41).妊娠週数,年齢により異なるが,NIPTによる出生前診断の陽性的中率は約54%,陰性的中率は99.99%以上と報告されている9).
約80%に先天性心疾患を合併する.ASD, VSD, PDAなどの単純型心疾患やTOFが多い40).13トリソミー27例の国内調査では,ASD 5例(22%),VSD 4例(17%),DORV 3例(13%)であった29).そのほか,TGA,大動脈縮窄(CoA),HLHS, AVSD,肺動脈弁狭窄(PS)も認められる40).18トリソミーに比して多弁病変の頻度は低い31).心疾患が新生児期の致死的合併症であることは少ない.
13トリソミーにおける肺高血圧の検討は少ないが,17.6~57%に合併すると報告されている29, 37).肺動脈の組織学的検討はほとんどないが,ASD,肺高血圧で肺動脈絞扼術を行った乳児の肺生検で,18トリソミーと同様に,肺小動脈中膜形成不全と内膜の線維性増生が認められた42).13トリソミーにおいても,先天的な肺小動脈中膜平滑筋の発達不良が示唆される.
近年,心内修復術後の生存例の報告も増加しており,米国の13トリソミーの心臓手術30症例の調査では,術後遠隔期の生存期間(中央値)は14.8年であった37).一方で,18トリソミーと同様に,単純型心疾患の手術については,非トリソミーの場合より約10倍死亡率が高いこと37),術前の人工呼吸管理を要した症例で術後の死亡率が高いと報告されている39).
性染色体異常の中で最も頻度が高く,胎児約70人に1人,出生女児2,000~2,500人に1人の頻度で認められる5, 43).低身長,性腺異形成による原発性無月経を来す.X染色体完全または部分欠失による.約50%がX染色体完全欠失(45,X)で,25%が45,X/46,XX, 45,X/47,XXXなどのモザイク,20%が同腕染色体や環状染色体などのX染色体の構造異常による44).欠失や構造異常は父の精子のX染色体由来が多い.父や母の加齢による発生頻度の上昇は明らかではない.X染色体のうちの1本は不活化されるため,通常X染色体上の遺伝子は1コピーの発現であるが,X染色体短腕末端(Xp22.3)の偽常染色体領域(pseudoautosomal region 1: PAR1)内の遺伝子は不活化を免れ,2コピー発現している.X染色体の欠失によりPAR1内の遺伝子群は1コピーに減少するため,それらの遺伝子の機能低下(ハプロ不全)がTurner症候群の原因と考えられている.PAR1上のSHOX遺伝子は軟骨の成長に関与するため,そのハプロ不全により低身長を呈する.
30~40%に心疾患を合併する.CoA(10%),BAV(30%),大動脈拡張(3~8%),大動脈弁狭窄(AS)(10%),HLHSなどの左心系閉塞性疾患が多い.ASDや部分肺静脈還流異常(主に左上肺静脈)もしばしば認められる43–46).X染色体短腕上のmatrix metalloproteinase組織阻害因子をコードするTIMP1遺伝子は,X染色体不活化を受けず,そのハプロ不全がTurner症候群に伴うBAV,大動脈拡張のリスクを増加させること,さらに機能的に類似したTIMP3遺伝子(22q12.3に座位)の変化が合併すると,そのリスクが10倍以上になると報告された44, 47).Turner症候群では,翼状頸などリンパ管低形成による徴候を伴うことから,リンパ流の阻害が左心系閉塞性疾患を招来している可能性があり,X染色体短腕上にリンパ管形成遺伝子の存在が推測された44).また,X染色体短腕上のKDM6A遺伝子は,遺伝子の転写制御にかかわるヒストン脱メチル化酵素をコードし,TIMP1遺伝子と同様にX染色体不活化を受けない.その機能低下は左心系閉塞性疾患の頻度が高い歌舞伎症候群の原因と考えられており,KDM6Aはリンパ管や左心系の発達に関与している可能性が示唆された44).
Turner症候群では,若年期から高血圧を合併しやすく,成人期に至るまでに約40%に認められ45, 48),冠動脈疾患,大動脈解離のリスクが高いとされている.高血圧の原因として,交感神経亢進,血管病変,内分泌異常など複数の因子が推測されている.定期的な血圧測定が必要であるが,Turner症候群では不安障害の合併が比較的多く,白衣高血圧がみられること,より治療が必要な夜間非降下型高血圧を鑑別する必要があることから,24時間自由行動下血圧測定(ABPM)の併用が推奨されている48).降圧治療にはβ遮断薬やアンジオテンシン受容体拮抗薬が用いられる48).また,3~8%に進行性の大動脈拡張が認められ,大動脈解離のリスクが増加する.通常大動脈解離は中年期以降に好発するが,Turner症候群においては中央値35歳で解離が認められたこと49),拡張がなくても解離が起こりうることが報告されており44),大動脈拡張のない症例であっても5年に1回の画像評価が推奨されている44, 45).Marfan症候群と同様に,大動脈解離部に嚢胞性中膜壊死が認められる場合があるが,有意な組織学的所見がないことも多く,Turner症候群における大動脈病変の機序は明らかにされていない49).
染色体22番長腕11.2領域の微細欠失に起因する.胎児1,000人に1人,出生4,000~5,000人に1人の頻度で認められ50),先天性心疾患においてはDown症候群についで頻度が高い染色体異常である.染色体検査(FISH法)は保険収載されている.
約90%は孤発例,約10%は家族例で,約70%が母子例である50, 51).父子例の報告が少ない理由として,男性患者の妊孕性が低いこと,男性の寿命が女性より短いこと,男性患者が所帯を担うことが困難であること,22q11.2欠失症候群患者の母親のほうが父親より染色体検査を受ける機会が多いことが考えられている52).
22q11.2領域の欠失は,約90%の症例で3Mbの共通領域であり(Fig. 2),約106の遺伝子が座位する.次いで,5~8%の症例で1.5Mbの領域の欠失が認められ,約50個の遺伝子が座位する.共通領域が欠失する理由として,22q11.2領域に存在する低頻度反復配列(low copy repeats: LCR)の関与が考えられている50, 51, 53–55).22q11.2内の5.6Mb領域に8個のLCR(A~H)があり,そのうち3Mb領域内の4個のLCR(A~D)が欠失に関与する.減数分裂時,通常は22番染色体アレル同士で同一配列が正対して相同組み換えが起こるが,2つのLCRが誤って付着して非アレル間で組み換えが起こると,LCR間を欠失した染色体と重複した染色体が形成される(Fig. 3a).重複染色体をもつ22q11.2重複症候群は,成長障害,精神発達遅滞,筋緊張低下などの非特異的な症状のため,実際より頻度が少なく見積もられている可能性がある56).22q11.2重複症候群85例の調査では,17例(20%)に先天性心疾患を認め,HLHS, VSD, TOF,総動脈幹(TrA),ASDなど多彩であった57).また,22番染色体内でも近接したLCRが付着して環状構造をとり,環状染色体とLCR間の領域を欠失した染色体が形成されることがある.環状染色体は不安定なため消失する(Fig. 3b).欠失が均一であるにもかかわらず,臨床症状は多様で,遺伝子型と表現型に相関は認められない.欠失も遺伝的背景も同一の一卵性双生児で表現型が大きく異なる症例が報告されており58),胎児循環における血流量の差など胎内環境の違いが関与することが示唆される.

Genes and LCR within 22q11.2 are shown. LCR, low copy repeats. (Modified from McDonald-McGinn et al50) and Morrow et al54))
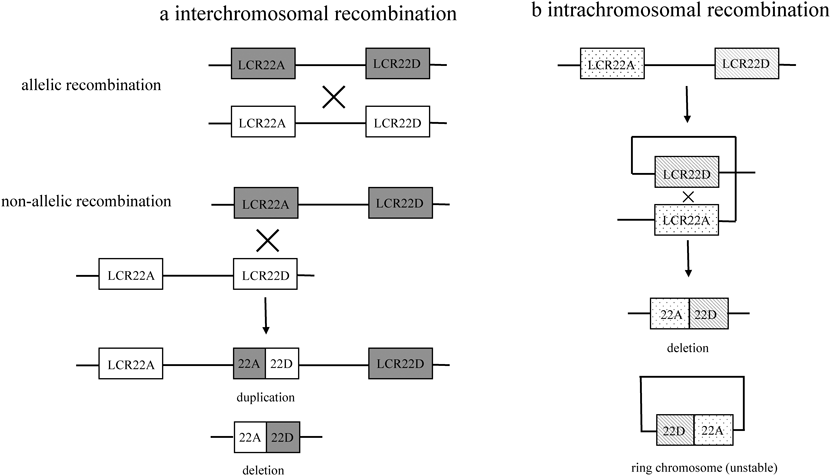
Rearrangements between LCR22A and LCR22D are shown for each chromosomal allele (white and gray). a) Interchromosomal recombination incorrectly occurs between LCR22A and LCR22D due to sequence similarity. “X” indicates crossover of two chromosomes resulting in duplication and deletion of genes flanked by LCR22A and LCR22D. b) Intrachromosomal recombination incorrectly occurs between proximal LCR22A (dotted) and distal LCR22D (shaded) forming a loop within one allele and resulting in a deletion and ring chromosome. The ring chromosome is unstable and unviable. LCR, low copy repeats. (Modified from McDonald-McGinn et al50))
22q11.2領域に存在する遺伝子のうち,T-ボックス型転写因子をコードするTBX1遺伝子は,胎生期の二次心臓領域(右心室,心臓流出路,心房に分化),原始咽頭弓(頭頸部器官,大動脈弓に分化)の細胞に発現し,背側の神経管から原始咽頭弓,心臓流出路に遊走する心臓神経堤細胞と相互作用して,胎生期にこれらの発達に関与する51, 59).そのほか,脳内の血管新生や骨格筋の分化,成人の前立腺癌の発生への関与も報告されている60–62).Tbx1ホモ接合性ノックアウトマウスで22q11.2欠失症候群の心疾患をはじめとする主要症状が観察されること,22q11.2欠失がなく,TBX1遺伝子のみの変異で22q11.2欠失症候群の症状を呈する症例があることから,TBX1は22q11.2欠失症候群の主要責任遺伝子とされている51, 53–55, 59).またTbx1発現を段階的に減少させたTbx1遺伝子量改変マウスの解析から,マウスでは,原始咽頭弓の発達はTbx1の遺伝子量依存的であることが示された63).
一方,TBX1変異があっても心疾患がない症例もあり,22q11.2欠失症候群の心疾患の原因はTBX1のハプロ不全のみではなく,22q11.2内外の遺伝子64)や環境因子の影響など複合的であることが考えられる.22q11.2領域に座位するDGCR6, UFD1L, TUPLE1, CRKL, COMT, PRODH, 22q11.2領域外のBMP4などの遺伝子や,mRNA分解に関与するmicroRNA, mRNA転写を制御するlong noncoding RNAの関与が示唆されている54, 55).また,ビタミンAの代謝産物であるレチノイン酸(RA)は,TBX1発現抑制作用を有し,Tbx1ノックアウトマウスにおいてRA産生を減少させると,心疾患が軽症化することが示されている65).近年,国内の91,664組の母子を対象とした大規模調査で,妊娠中のビタミンAの過剰摂取が,先天性心疾患の発症リスクを5倍以上上昇させることが報告され66),RAと先天性心疾患の関連が疫学研究からも示唆された.また,妊娠糖尿病により,胎児に22q11.2欠失症候群様の心血管表現型を呈することが知られており,高血糖が咽頭弓組織でのRA上昇を介してTBX1発現を低下させる機序が推測された55).以上のように,22q11.2欠失症候群の発症にはTBX1をはじめとする遺伝的要因に加えて,環境要因の関与も大きいことが考えられる.
円錐動脈幹部(心臓流出路,特に右室流出路)と大動脈弓の異常を特徴とする.Table 3に22q11.2欠失症候群で頻度の高い心疾患を示す.TOF, VSD, B型IAAやTrAが特徴的であり,心内病変のない孤立性大動脈弓異常も認められる45, 53, 67–69).TOFの重症型である肺動脈閉鎖兼心室中隔欠損(PAVSD)の合併も10~25%に認められ,肺血流の供給血管として,動脈管よりも主要体肺側副動脈(MAPCA)が主であることが多い67, 68, 70).22q11.2欠失症候群において,胎生期に円錐動脈幹の発達が障害され,右室流出路が低形成になると,背側大動脈から分枝する節間動脈が体肺交通を保って,MAPCAとして存続する.MAPCAは肺血流過多,減少,区域性肺高血圧,気管支軟化症など,多様な症状,血行動態異常の原因となる71, 72).
| CHD | Frequency in 22q11.2 deletion syndrome |
|---|---|
| TOF | 18–40% |
| VSD | 10–25% |
| PAVSD with PDA | 2–10% |
| PAVSD with MAPCA | 20–25% |
| Aortic arch anomaly | 10–15% |
| IAA | 5–15% |
| TrA | 2–10% |
| CHD, congenital heart disease; IAA, interruption of aortic arch; MAPCA, major aortopulmonary collateral arteries; PAVSD, pulmonary atresia with ventricular septal defect; PDA, patent ductus arteriosus; TOF, tetralogy of Fallot; TrA, truncus arteriosus; VSD, ventricular septal defect. (Modified from Pierpont et al45), McDonald-McGinn et al53), Momma67), Unolt et al68), and Campbell et al69)) | |
Table 4に,22q11.2欠失の頻度が高い先天性心疾患を示す.IAA, PAVSD MAPCA, TrAにおいて欠失頻度が高く,22q11.2欠失症候群に特徴的な心疾患であるといえる67, 70, 73–75).機序は不明であるが,IAAで欠失が認められる症例はほぼB型で,A型における欠失はまれである67, 73, 74).TrAでは,Van Praagh分類A1型からA4型のすべてのタイプで欠失を認めるが73),左右肺動脈の連続性が欠如しているA3型で,特に欠失の頻度が高いと報告されている67, 68, 73, 74).一方で,A4型TrAは,22q11.2欠失症候群に特徴的なB型IAAとTrAが合併した形態であるが,他のタイプより欠失の頻度が高いという報告はない73).また,流出路型VSDで欠失の頻度が高いという報告が複数あり45, 68, 73, 74),22q11.2欠失症候群に特徴的な円錐動脈幹異常と合致するが,VSDの位置と欠失の頻度には相関がなかったという報告67, 76),あるいは流出路型VSDでは欠失が認められなかったという報告77)もある.右大動脈弓,鎖骨下動脈起始異常などの大動脈弓異常を合併しているVSDでは,VSDの位置によらず22q11.2欠失の頻度が高くなるとされている67, 68, 73–76).また,心内病変を伴わない孤立性大動脈弓異常における欠失の頻度は報告により5~24%と幅があるが73, 78),心内病変を合併する大動脈弓異常では欠失の頻度が増加する67, 73).
| CHD | frequency |
|---|---|
| TOF | 5–15% |
| PAVSD PDA | 10–20% |
| PAVSD MAPCA | 40–60% |
| IAA | 50–89% |
| TrA | 12–40% |
| VSD | 2–15% |
| Aortic arch anomaly | 5–24% |
| DORV | 1–2% |
| TGA | 1% |
| CHD, congenital heart disease; DORV, double outlet right ventricle; IAA, interruption of aortic arch; MAPCA, major aortopulmonary collateral arteries; PAVSD, pulmonary atresia with ventricular septal defect; PDA, patent ductus arteriosus; TGA, transposition of great arteries; TOF, tetralogy of Fallot; TrA, truncus arteriosus; VSD, ventricular septal defect. (Modified from Momma67), Maeda et al70), Peyvandi et al74), Agergaard et al75), and McElhinney et al78)) | |
以上に述べたように,TOF,PAVSD MAPCA,B型IAA,TrA,大動脈弓異常を伴うVSDでは,22q11.2欠失の頻度が高いため,心外合併症の有無を問わず,染色体FISH法による欠失のスクリーニングを行うことが推奨されている45).特徴的な顔貌はほとんどの22q11.2欠失症候群に合併するが,新生児,乳児期早期や成人期には明確でない場合も多く,心疾患を発端に染色体検査を行った結果,22q11.2欠失症候群の診断に至ることがある.欠失の診断により,心外合併症の評価,管理を早期から開始でき,包括的な健康管理に結びつけることができるメリットがある50, 51).
TOFやTrAの発症機序として,二次心臓領域細胞が形成する円錐動脈幹,特に主肺動脈から肺動脈弁下漏斗部にかけての低形成,無形成が提唱されている59, 79).マウスにおいてTbx1発現細胞の挙動を経時的に追跡した解析で,二次心臓領域に発現するTbx1細胞は,肺動脈弁下漏斗部,主肺動脈へ分化することが明らかにされ,TBX1機能が低下する22q11.2欠失症候群においてTOFやTrAの頻度が高いことと一致する68, 80).一方,同様に円錐動脈幹異常であるDORVやTGAにおける22q11.2欠失の頻度は低く67, 68, 74),円錐動脈幹の左方移動の障害に起因するDORVや,回旋の障害に起因するTGAでは,TBX1以外の分子経路の関与が示唆された59, 79).
染色体7番長腕11.23領域の微細欠失に起因する.欠失領域(1.5~1.8Mb)にある遺伝子のハプロ不全と考えられる.7,500人に1人の頻度で認められる.孤発例がほとんどで,家族例の報告は少ない81, 82).同じ欠失を有していても,表現型は多様である81).染色体検査(FISH法)は保険収載されている.
22q11.2欠失症候群と同様に,欠失領域の両端に3個のLCR(A, B, C)が存在し,LCR間の非相同染色体組み換えにより,均一な欠失が高率に起こると推測されている.共通の欠失領域は,Williams syndrome critical region(WSCR)と呼ばれ,25~27個の遺伝子が座位する.幼児期の妖精様顔貌,精神発達遅滞,特異な性格,大動脈弁上部狭窄(SVAS),末梢性肺動脈狭窄(PPS)などの心血管病変,乳児期の高カルシウム血症など特徴的な症状,所見を呈する症候群である.組み換えの過程で欠失と同様に生じる7q11.23重複症候群は,発達遅滞をはじめとする非特異的な症状に加えて,機序は不明であるが,Williams症候群と対照的な大動脈拡大を呈することが報告されている83, 84).
WSCR内に座位するELN遺伝子の産物であるエラスチンは,弾性線維の主要構成成分で,肺,皮膚,大動脈などの弾性動脈に存在し,組織の伸展と弛緩,弾性に関与する.Williams症候群の心外症状をもたない家族性SVAS家系において,ELN遺伝子の変異が報告されていること85, 86),SVAS症例でELN遺伝子の一塩基欠失や置換が同定されたこと87)から,ELN遺伝子は,本症候群におけるSVASの疾患責任遺伝子と考えられる.一方で,Elnヘテロ接合性ノックアウトマウスでは,全身の動脈中膜肥厚,高血圧,心肥大が認められるが,SVASは明らかでない82).
エラスチン異常による全身の動脈狭窄性病変を生じる.SVAS, PPSのほか,冠動脈,腎血管,脳血管の狭窄の合併も知られている.エラスチンの機能低下により血管壁の弾力性が低下し,壁でのずり応力が上昇すること,エラスチンによる血管平滑筋増殖の調節機能が低下することが原因で,血管内膜,中膜が肥厚し,内腔の狭窄を生じると考えられている83).Williams症候群に特徴的な心大血管病変であるSVAS, PPSの自然歴を10年以上にわたり追跡した調査では,多くのSVAS症例で経年的に狭窄が進行したが,圧較差20 mmHg以下の軽症例では,進行が認められなかった.また,大動脈低形成を伴うSVAS症例では,大動脈形成術後も再狭窄が進行した.一方,PPSについては,圧較差30 mmHg以上であっても,ほとんどの症例で自然軽快した88).PPSが経年的に改善する理由は明らかにされていない.
Williams症候群で注意すべき動脈狭窄病変として,冠動脈狭窄があり,5~9%に認められる89).SVASによる乱流が冠動脈入口部にずり応力を及ぼし,狭窄の進行に関与することが推測されるが,SVASがなくても起こりうる83).冠動脈狭窄は突然死の原因となり90, 91),一般人口に比して25~100倍突然死のリスクが高いと報告されている91).また,全身麻酔が心停止と関連し,3歳未満,流出路狭窄の症例で特にリスクが高いが92),狭窄病変が軽度でも起こり得る82).Williams症候群では,SVASによる左室肥大,エラスチン欠乏による拡張期の大動脈弾性低下,酸素需要増大,冠動脈狭窄など心筋虚血を誘発する複合的な要因がある.全身麻酔による体血管抵抗低下,心機能低下が加わると,さらに冠血流が低下して重篤な心筋虚血を来す可能性がある.このため,全身麻酔時には血圧を急に低下させない管理が望ましいとされている82, 92).
高血圧はWillimas症候群の40~50%に合併し,小児期からもしばしば認められる81–83).その原因として,エラスチン欠乏による動脈弾性低下が考えられているが81),Willimas症候群の50%に腎動脈狭窄が合併することから,腎血管性高血圧の関与も推測される.また,WSCRのテロメア側に座位し,活性酸素産生に関与するNCF1遺伝子の機能低下が,高血圧を軽症化させる可能性が報告された93).
1番染色体短腕末端の1p36領域の欠失に起因する.5,000~10,000人に1人の頻度で認められる.95%は孤発例で,欠失を生じる1番染色体は母由来が60%,父由来が40%とされている94).染色体検査(FISH法)は保険収載され,検査会社で,1p36の共通欠失領域を含むプローブを用いて行われている.マイクロアレイを用いた染色体検査であるアレイCGH(comparative genomic hybridization)が普及した結果,原因不明の精神発達遅滞患者の遺伝学的スクリーニング検査が多く行われようになり,1p36欠失の同定,および本症候群の理解が進んだ94).特徴的な顔貌(小頭,短頭,落ちくぼんだ眼,尖ったオトガイ),成長障害,精神発達遅滞,てんかん,先天性心疾患を来す94–96).
約40~70%に先天性心疾患を認め,VSD, PDA, ASDなどの単純型心疾患が多い.また,1p36欠失症候群の特徴として,約10~20%に左室心筋緻密化障害,拡張型心筋症などの心筋症を合併する94–97).従来欠失のサイズと臨床症状との相関は乏しいとされてきたが94, 95),近年1p36遠位部欠失が,近位部欠失と比較して心疾患を合併する頻度が有意に高いことが明らかになった96).また,遠位部欠失領域(1p36.32)に座位し,ミトコンドリア機能調節に関与するPRDM16遺伝子の機能喪失が左室心筋緻密化障害,拡張型心筋症の発症に関与することが報告されたが98),PRDM16変異があっても心筋症のない症例も多く,不完全浸透が示唆される.そのほか,VSDやASDの原因として,遠位部欠失領域(1p36.23)に座位するRERE遺伝子,近位部欠失領域(1p36.12)に座位するECE1遺伝子が候補に挙げられている96).
染色体4番短腕の部分欠失に起因し,4p16.3の1.5Mb責任領域(Wolf-Hirschhorn syndrome critical region: WHSCR)の欠失による遺伝子群のハプロ不全により発症する.出生20,000から50,000人に1人の頻度で認められる.男女比は1 : 2で,女児に多い.50~60%は4p端部の単純欠失であり,欠失の大きさは様々で,85%以上が孤発例である.約15%で不均衡型転座を認め,片親が均衡型転座保因者である可能性がある.欠失を生じる4番染色体は80%が父由来とされている99, 100).G分染法による欠失の同定は50~60%であり,確定診断には,4p16のWHSCRを含むプローブを用いたFISH法が行われる100).FISH法は保険収載され,検査会社で行われている.端部側の責任領域(WHSCR-2)の存在も報告されている101, 102).
ギリシャ兵士ヘルメット様といわれる特徴的な顔貌を呈する.成長障害,精神発達遅滞,難治性てんかんを高率に認める.てんかん発作の頻度は高く,3歳までに90%以上に発作を認めるとされている100, 102).4p16.3内に0.54Mbのてんかん責任領域が存在する可能性が報告された102).
50~86%に先天性心疾患を合併し,ASD, VSD, PDA, PSなど単純型心疾患が多い.国内22例の検討では,ASDが13例(59%)と最も多く,PS 6例,PDA 5例で,複雑型心疾患は認められなかった.欠失の大きさや部位と心疾患の程度に相関はなく,心疾患責任領域は同定されていない102).また,高コレステロール血症を36%に認めたと報告されており,動脈硬化予防のため定期的な血液検査が望ましい102).
染色体5番短腕の部分欠失に起因する.出生15,000から50,000人に1人の頻度で認められる.80~90%は5p端部の単純欠失であり,欠失の大きさは様々で,約85%は孤発例である.約15%は不均衡型転座で,片親が均衡型転座保因者である可能性がある.欠失を生じる5番染色体は約80%が父由来とされている103, 104).G分染法による染色体検査では欠失が同定されないことがある.5pプローブを用いた染色体検査(FISH法)は一般的に行われていないが,マイクロアレイ染色体検査で染色体ゲノムのコピー数変化を検出することで診断可能である.マイクロアレイ検査は保険収載され,検査会社で行われている.
小頭,成長障害,精神発達遅滞,筋緊張低下,特徴的な顔貌を呈する.乳児期の哺乳障害,側弯の頻度が高い103).出生後の猫が鳴くような甲高い鳴き声が診断の契機となる.鳴き声の性状から,従来猫鳴き症候群(Cri du Chat syndrome)と呼称されていたが,動物名が入った病名は不適切とされ,現在は使用されなくなっている.甲高い鳴き声の責任領域は5p13.3と推測されている103).
約30%に先天性心疾患を合併し,VSD, ASD, PDAなどの単純型心疾患が多い103).心疾患の責任領域として,5p中間部の3.6Mb領域が候補として報告された104, 105).
17番染色体短腕(17p11.2)の中間部欠失,または同領域に座位するRAI1(retinoic acid induced 1)遺伝子変異によるハプロ不全に起因する.出生15,000~20,000人に1人に認められる.大半が孤発例である.17p11.2領域には約80の遺伝子が座位し,90%はRAI1遺伝子を含む17p11.2領域の欠失で,10%はRAI1遺伝子変異で発症する106, 107).欠失範囲は,1.5~9Mbと幅広いが,70~80%の症例が3.7Mbの共通欠失領域を有する.22q11.2欠失症候群やWilliams症候群と同様に,3.7Mb領域の両端にあるLCR間で非相同染色体組み換えが起き,均一な欠失が起こると推測されている107, 108).欠失はG分染法では検出できないことが多い.17p11.2領域プローブを用いたFISH法は一般的に行われていないが,検査会社で行われているマイクロアレイ染色体検査で欠失の検出が可能であり,保険収載されている.
特徴的顔貌(短頭,幅広い前額,短い人中,テント状の上口唇,太い眉毛),短指症,低身長,肥満,精神発達遅滞のほか,睡眠障害(夜間,明け方に覚醒,日中の眠気),自傷行為,身体の穴に物を入れる行為,感情爆発という特徴的な症状がある.これらの症状は1歳6か月以降に明らかになることが多い108, 109).睡眠障害の原因は,睡眠を促進するメラトニン分泌の日内リズムの逆転とされてきたが,日内リズム異常のない症例でも認められることから,他の要因の関与も考えられている108, 110).RAI1は脳に多く発現するクロマチン結合蛋白で,細胞増殖や細胞周期に関わる遺伝子の発現調節に関与し,中枢神経や日内リズムの発達を制御する108).
25~45%に先天性心疾患を合併し,ASD, VSD, PS, TOFの頻度が高い108, 111).12%に不整脈を認めたという報告がある111).心疾患の種類や重症度と欠失範囲の間には明らかな相関は認められていない.Smith-Magenis症候群24例について,心エコーによる心機能解析を行った検討では,左室拡張能の低下が認められ,潜在的な心機能障害が示唆された111).また,心疾患の合併は17p11.2欠失症例で認められ,RAI1遺伝子変異症例では報告されていない111).
先天性心疾患を合併する頻度の高い染色体異常症候群で,染色体検査が保険収載されている疾患を取り上げ,遺伝的背景,心疾患の特徴を中心に述べた.心疾患,あるいは心外疾患も含めた診断を契機に染色体異常症候群が明らかとなることで,合併症の早期診断,治療,包括的な健康管理が可能となり,ひいては患者の予後改善,生活の質の向上につながる可能性がある.そのためには,先天性心疾患を診断,治療する小児循環器科,心臓血管外科に加えて,臨床遺伝科をはじめとする関連診療科およびコメディカルとの共同体制による診療が不可欠である.染色体異常症候群の診断を端緒に,先天性心疾患発症のメカニズムが明らかになり,予防と治療法の開発につながることを期待したい.
英文を校正いただいた,東京都立小児総合医療センター臨床研究支援センターValera James Robert先生に深謝いたします.
本論文に関して開示すべき利益相反はない.
1) 日本小児循環器学会:小児期発症心疾患実態調査2021集計結果報告書.2022年11月 https://jspccs.jp/wp-content/uploads/JSPCCSNewsLetter2022-3.pdf
2) Yasuhara J, Garg V: Genetics of congenital heart disease: A narrative review of recent advances and clinical implications. Transl Pediatr 2021; 10: 2366–2386
3) Landstrom AP, Kim JJ, Gelb BD, et al: American Heart Association Council on Genomic and Precision Medicine; Council on Lifelong Congenital Heart Disease and Heart Health in the Young; Council on Arteriosclerosis, Thrombosis and Vascular Biology; and Council on Lifestyle and Cardiometabolic Health: Genetic testing for heritable cardiovascular diseases in pediatric patients: A scientific statement from the American Heart Association. Circ Genom Precis Med 2021; 14: e000086
4) Kodo K, Uchida K, Yamagishi H: Genetic and cellular interaction during cardiovascular development implicated in congenital heart diseases. Front Cardiovasc Med 2021; 8: 653244
5) Nussbaum RL, McInnes RR, Willard HF (eds): Principals of clinical cytogenetics and genome analysis, in Thompson & Thompson Genetics in Medicine 8th eds, Philadelphia, Elsevier, 2016, pp57–74
6) 上砂光裕:小児循環器の遺伝学的検査—小児循環器領域の生涯包括遺伝医療—.小児科診療2019; 82: 841–846
7) Jones KL, Jones MC, Campo MD: Down syndrome, in Jones KL, Jones MC, Campo MD (eds): Smith’s Recognizable Patterns of Human Malformation, 8th eds, Philadelphia, Elsevier, 2022, pp1–7
8) 循環器病の診断と治療に関するガイドライン(2010年度合同研究班報告):染色体異常,心臓血管疾患における遺伝学的検査と遺伝カウンセリングに関するガイドライン(2011年改訂版), 2011, pp. 13–24. https://www.j-circ.or.jp/cms/wp-content/uploads/2020/02/JCS2011_nagai_h.pdf
9) 鈴森伸宏:母体血を用いた非侵襲性出生前遺伝学的検査(NIPT)の現状と展望.周産期医学2022; 52: 651–654
10) Truong DT, Minich LL, Maleszewski JJ, et al: Atrioventricular septal defects, in Robert ES, Penny DJ, Feltes TF, et al. (eds): Moss and Adams’ Heart Disease in Infants, Children and Adolescents, 10th eds, Philadelphia, Wolters Kluwer, 2022, pp721–745
11) Bull MJ: Down syndrome. N Engl J Med 2020; 382: 2344–2352
12) Bergström S, Carr H, Petersson G, et al: Trends in congenital heart defects in infants with Down syndrome. Pediatrics 2016; 138: e20160123
13) 小穴慎二,市田蕗子,太田八千緒,ほか:平成14~16年度研究課題報告Down症候群の心血管疾患—核型と表現型,肺高血圧に関する検討—.日小児循環器会誌2010; 26: 58–68
14) Scala C, Maggiore ULR, Candiani M, et al: Aberrant right subclavian artery in fetuses with Down syndrome: A systematic review and meta-analysis. Ultrasound Obstet Gynecol 2015; 46: 266–276
15) 北野正尚,杉山 央,矢内 淳,ほか:ダウン症候群における大動脈弓形態の特徴.日小児循環器会誌2001; 17: 420–423
16) Asim A, Agarwal S, Panigrahi I, et al: CRELD1 gene variants and atrioventricular septal defects in Down syndrome. Gene 2018; 641: 180–185
17) Dimopoulos K, Constantine A, Clift P, et al: for Down Syndrome International (DSi): Cardiovascular complications of Down syndrome: Scoping review and expert consensus. Circulation 2023; 147: 425–441
18) Nguyen HH, Jay PY: A single misstep in cardiac development explains the co-occurrence of Tetralogy of Fallot and complete atrioventricular septal defect in Down syndrome. J Pediatr 2014; 165: 194–196
19) Saji T: Clinical characteristics of pulmonary arterial hypertension associated with Down syndrome. Pediatr Int 2014; 56: 297–303
20) Fukushima H, Kosaki K, Sato R, et al: Mechanisms underlying early development of pulmonary vascular obstructive disease in Down syndrome: An imbalance in biosynthesis of thromboxane A2 and prostacyclin. Am J Med Genet A 2010; 152A: 1919–1924
21) Galambos C, Minic AD, Bush D, et al: Increased lung expression of anti-angiogenic factors in Down syndrome: Potential role in abnormal lung vascular growth and the risk for pulmonary hypertension. PLoS One 2016; 11: e0159005
22) Iwaya Y, Muneuchi J, Inoue Y, et al: Relationship between pulmonary arterial resistance and compliance in patients with Down syndrome. Pediatr Cardiol 2019; 40: 841–847
23) Masaki N, Saiki Y, Endo M, et al: Is trisomy 21 a risk factor for rapid progression of pulmonary arteriopathy?: Revisiting histopathological characteristics using 282 lung biopsy specimens. Circ J 2018; 82: 1682–1687
24) Masaki N, Saiki Y, Endo M, et al: Evidence of pulmonary vascular reverse remodeling after pulmonary artery banding performed in early infancy in patients with congenital heart defects. Circ J 2018; 82: 684–690
25) Hoashi T, Hirahara N, Murakami A, et al: Current surgical outcomes of congenital heart surgery for patients with Down syndrome in Japan. Circ J 2018; 82: 403–408
26) Sarno LA, Walters HL 3rd, Bondarenko I, et al: Significant improvements in mortality after the Fontan operation in children with Down syndrome. Ann Thorac Surg 2020; 109: 835–841
27) Jones KL, Jones MC, Campo MD: Trisomy 18 syndrome, in Jones KL, Jones MC, Campo MD (eds): Smith’s Recognizable Patterns of Human Malformation, 8th eds, Philadelphia, Elsevier, 2022, pp8–13
28) Kepple JW, Fishler KP, Peeples ES: Surveillance guidelines for children with trisomy 18. Am J Med Genet A 2021; 185: 1294–1303
29) Maeda J, Yamagishi H, Furutani Y, et al: The impact of cardiac surgery in patients with trisomy 18 and trisomy 13 in Japan. Am J Med Genet A 2011; 155A: 2641–2646
30) Van Praagh S, Truman T, Firpo A, et al: Cardiac malformations in trisomy 18: A study of 41 postmortem cases. J Am Coll Cardiol 1989; 13: 1586–1597
31) 片岡功一:18トリソミーおよび13トリソミー児の心臓血管手術.日小児循環器会誌2020; 36: 3–15
32) Tahara M, Sanada K, Morita R, et al: Insufficient development of vessels and alveoli in lungs of infants with trisomy 18-Features of pulmonary histopathological findings from lung biopsy. Am J Med Genet A 2021; 185: 1059–1066
33) Yamaki S: Pulmonary vascular disease associated with pulmonary hypertension in 445 patients: Diagnosis from lung biopsy and autopsy. Gen Thorac Cardiovasc Surg 2013; 61: 24–31
34) 田原昌博:18トリソミー—肺生検組織を中心に—.日小児循環器会誌2023; 39: 51–61
35) Hatai E, Muneuchi J, Sugitani Y, et al: Pulmonary vascular resistance and compliance in individuals with trisomy 18. Am J Med Genet A 2022; 188: 534–539
36) Czosek RJ, Baskar S, Mohan S, et al: Incidence and outcome of arrhythmias and electrical disease in patients with trisomy 18. Am J Med Genet A 2023; 191: 2518–2523
37) Peterson JK, Kochilas LK, Catton KG, et al: Long-term outcomes of children with trisomy 13 and trisomy 18 after congenital heart disease interventions. Ann Thorac Surg 2017; 103: 1941–1949
38) Nakai R, Fujioka T, Okamura K, et al: Survival outcomes of two-stage intracardiac repair in large ventricular septal defect and trisomy 18. Pediatr Cardiol 2021; 42: 821–831
39) Cooper DS, Riggs KW, Zafar F, et al: Cardiac surgery in patients with trisomy 13 and 18: An analysis of the society of thoracic surgeons congenital heart surgery database. J Am Heart Assoc 2019; 8: e012349
40) Kepple JW, Fishler KP, Peeples ES: Surveillance guidelines for children with trisomy 13. Am J Med Genet A 2021; 185: 1631–1637
41) Jones KL, Jones MC, Campo MD: Trisomy 13 syndrome, in Jones KL, Jones MC, Campo MD (eds): Smith’s Recognizable Patterns of Human Malformation, 8th eds, Philadelphia, Elsevier, 2022, pp14–17
42) Tahara M, Shimozono S, Nitta T, et al: Medial defects of the small pulmonary arteries in fatal pulmonary hypertension in Infants with trisomy 13 and trisomy 18. Am J Med Genet A 2014; 164A: 319–323
43) Jones KL, Jones MC, Campo MD: 45X syndrome, in Jones KL, Jones MC, Campo MD (eds): Smith’s Recognizable Patterns of Human Malformation, 8th eds, Philadelphia, Elsevier, 2022, pp78–83
44) Gravholt CH, Viuff MH, Brun S, et al: Turner syndrome: Mechanisms and management. Nat Rev Endocrinol 2019; 15: 601–614
45) Pierpont ME, Brueckner M, Chung WK, et al: American Heart Association Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; and Council on Genomic and Precision Medicine: Genetic basis for congenital heart disease: Revisited: A scientific statement from the American Heart Association. Circulation 2018; 138: e653–e711
46) Mittal S, Breckpot J: Genetic aspects of congenital heart defects, in Robert ES, Penny DJ, Feltes TF, et al (eds): Moss and Adams’ Heart Disease in Infants, Children and Adolescents, 10th eds, Philadelphia, Wolters Kluwer, 2022, pp84–112.
47) Corbitt H, Morris SA, Gravholt CH, et al: GenTAC Registry Investigators: TIMP3 and TIMP1 are risk genes for bicuspid aortic valve and aortopathy in Turner syndrome. PLoS Genet 2018; 14: e1007692
48) Jones L, Blair J, Hawcutt DB, et al: Hypertension in Turner syndrome: A review of proposed mechanisms, management and new directions. J Hypertens 2023; 41: 203–211
49) Gravholt CH, Landin-Wilhelmsen K, Stochholm K, et al: Clinical and epidemiological description of aortic dissection in Turner’s syndrome. Cardiol Young 2006; 16: 430–436
50) McDonald-McGinn DM, Sullivan KE, Marino B, et al: 22q11.2 deletion syndrome. Nat Rev Dis Primers 2015; 1: 15071
51) Yamagishi H: The 22q11.2 deletion syndrome. Keio J Med 2002; 51: 77–88
52) Costain G, Chow EWC, Silversides CK, et al: Sex differences in reproductive fitness contribute to preferential maternal transmission of 22q11.2 deletions. J Med Genet 2011; 48: 819–824
53) McDonald-McGinn DM, Hain HS, Emanuel BS, et al: 22q11.2 Deletion Syndrome. GeneReviews®. https://www.ncbi.nlm.nih.gov/books/NBK1116/#IX-22
54) Morrow BE, McDonald-McGinn DM, Emanuel BS, et al: Molecular genetics of 22q11.2 deletion syndrome. Am J Med Genet A 2018; 176: 2070–2081
55) Du Q, de la Morena MT, van Oers NSC: M de la Morena T, van Oers NSC: The genetics and epigenetics of 22q11.2 deletion syndrome. Front Genet 2020; 10: 1365
56) Bartik LE, Hughes SS, Tracy M, et al: 22q11.2 duplications: Expanding the clinical presentation. Am J Med Genet A 2022; 188: 779–787
57) Butensky A, de Rinaldis CP, Patel S, et al: Cardiac evaluation of patients with 22q11.2 duplication syndrome. Am J Med Genet A 2021; 185: 753–758
58) Yamagishi H, Ishii C, Maeda J, et al: Phenotypic discordance in monozygotic twins with 22q11.2 deletion. Am J Med Genet 1998; 78: 319–321
59) 山岸敬幸:先天性心疾患の理解を深めるMolecular Embryology.日小児循環器会誌2007; 23: 364–372
60) Cioffi S, Martucciello S, Fulcoli FG, et al: Tbx1 regulates brain vascularization. Hum Mol Genet 2014; 23: 78–89
61) Cen H, Luo H, Luo B, et al: TBX1 regulates myogenic differentiation by activating the TGFβ-Smad2/3 pathway in myoblasts. Exp Biol Med (Maywood) 2023; 248: 61–69
62) Cui J, Zhang Y, Ren X, et al: TBX1 functions as a tumor activated in prostate cancer by promoting ribosome RNA gene transcription. Front Oncol 2021; 10: 616173
63) Hu T, Yamagishi H, Maeda J, et al: Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development 2004; 131: 5491–5450
64) Yamagishi H, Maeda J, Hu T, et al: Tbx1 is regulated by tissue-specific forkhead proteins through a common Sonic hedgehog-responsive enhancer. Genes Dev 2003; 17: 269–281
65) Voss AK, Vanyai HK, Collin C, et al: MOZ regulates the Tbx1 Locus, and Moz mutation partially phenocopies DiGeorge syndrome. Dev Cell 2012; 23: 652–663
66) Kawai S, Pak K, Iwamoto S, et al: Japan Environment and Children’s Study Group: Association between maternal factors in early pregnancy and congenital heart defects in offspring: The Japan Environment and Children’s Study. J Am Heart Assoc 2023; 12: e029268
67) Momma K: Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome. Am J Cardiol 2010; 105: 1617–1624
68) Unolt M, Versacci P, Anaclerio S, et al: Congenital heart diseases and cardiovascular abnormalities in 22q11.2 deletion syndrome: From well-established knowledge to new frontiers. Am J Med Genet A 2018; 176: 2087–2098
69) Campbell IM, Sheppard SE, Crowley TB, et al: What is new with 22q?: An update from the 22q and You Center at the Children’s Hospital of Philadelphia. Am J Med Genet A 2018; 176: 2058–2069
70) Maeda J, Yamagishi H, Matsuoka R, et al: Frequent association of 22q11.2 deletion with tetralogy of Fallot. Am J Med Genet 2000; 92: 269–272
71) Yamagishi H, Maeda J, Higuchi M, et al: Bronchomalacia associated with pulmonary atresia, ventricular septal defect and major aortopulmonary collateral arteries, and chromosome 22q11.2 deletion. Clin Genet 2002; 62: 214–219
72) Ishida H, Maeda J, Uchida K, et al: Unique pulmonary hypertensive vascular diseases associated with heart and lung developmental defects. J Cardiovasc Dev Dis 2023; 10: 333
73) Goldmuntz E: 22q11.2 deletion syndrome and congenital heart disease. Am J Med Genet C Semin Med Genet 2020; 184: 64–72
74) Peyvandi S, Lupo PJ, Garbarini J, et al: 22q11.2 deletions in patients with conotruncal defects: Data from 1,610 consecutive cases. Pediatr Cardiol 2013; 34: 1687–1694
75) Agergaard P, Olesen C, Østergaard JR, et al: The prevalence of chromosome 22q11.2 deletions in 2,478 children with cardiovascular malformations: A population-based study. Am J Med Genet A 2012; 158A: 498–508
76) McElhinney DB, Driscoll DA, Levin ER, et al: Chromosome 22q11 deletion in patients with ventricular septal defect: Frequency and associated cardiovascular anomalies. Pediatrics 2003; 112: e472–e476
77) Yamagishi H, Maeda J, Tokumura M, et al: Ventricular septal defect associated with microdeletions of chromosome 22q11.2. Clin Genet 2000; 58: 493–496
78) McElhinney DB, Clark BJ 3rd, Weinberg PM, et al: Association of chromosome 22q11 deletion with isolated anomalies of aortic arch laterality and branching. J Am Coll Cardiol 2001; 37: 2114–2119
79) 山岸敬幸,山岸千尋:流出路の発生とその異常,山岸敬幸,白石 公(編):新先天性心疾患を理解するための臨床心臓発生学.東京,MEDICAL VIEW, 2021, pp185–194
80) Maeda J, Yamagishi H, McAnally J, et al: Tbx1 is regulated by forkhead proteins in the secondary heart field. Dev Dyn 2006; 235: 701–710
81) Morris CA: Williams syndrome. GeneReview®. https://www.ncbi.nlm.nih.gov/books/NBK1249/
82) Kozel BA, Barak B, Kim CA, et al: Williams syndrome. Nat Rev Dis Primers 2021; 7: 42
83) Pober BR: Williams-Beuren syndrome. N Engl J Med 2010; 362: 239–252
84) Parrott A, James J, Goldenberg P, et al: Aortopathy in the 7q11.23 microduplication syndrome. Am J Med Genet A 2015; 167A: 363–370
85) Micale L, Turturo MG, Fusco C, et al: Identification and characterization of seven novel mutations of elastin gene in cohort of patients affected by supravalvular aortic stenosis. Eur J Hum Genet 2010; 18: 317–323
86) Hayano S, Okuno Y, Tsutsumi M, et al: Frequent intragenic microdeletions of elastin in familial supravalvular aortic stenosis. Int J Cardiol 2019; 274: 290–295
87) Li DY, Toland AE, Boak BB, et al: Elastin point mutations cause an obstructive vascular disease, supravalvular aortic stenosis. Hum Mol Genet 1997; 6: 1021–1028
88) Wessel A, Pankau R, Kececioglu D, et al: Three decades of follow-up of aortic and pulmonary vascular lesions in the Williams-Beuren syndrome. Am J Med Genet 1994; 52: 297–301
89) Matisoff AJ, Olivieri L, Schwartz JM, et al: Risk assessment and anesthetic management of patients with Williams syndrome: A comprehensive review. Paediatr Anaesth 2015; 25: 1207–1215
90) Bird LM, Billman GF, Lacro RV, et al: Sudden death in Williams syndrome: Report of ten cases. J Pediatr 1996; 129: 926–931
91) Wessel A, Gravenhorst V, Buchhorn R, et al: Risk of sudden death in the Williams-Beuren syndrome. Am J Med Genet A 2004; 127A: 234–237
92) Latham GJ, Ross FJ, Eisses MJ, et al: Perioperative morbidity in children with elastin arteriopathy. Paediatr Anaesth 2016; 26: 926–935
93) Kozel BA, Danback JR, Williams JL, et al: Williams syndrome predisposes to vascular stiffness modified by antihypertensive use and copy number changes in NCF1. Hypertension 2014; 63: 74–79
94) Gajecka M, Mackay KL, Shaffer LG: Monosomy 1p36 deletion syndrome. Am J Med Genet C Semin Med Genet 2007; 145: 346–356
95) Jones KL, Jones MC, Campo MD: 1p36 deletion syndrome, in Jones KL, Jones MC, Campo MD (eds): Smith’s Recognizable Patterns of Human Malformation, 8th eds, Philadelphia, Elsevier, 2022, pp76–79
96) Jacquin C, Landais E, Poirsier C, et al: 1p36 deletion syndrome: Review and mapping with further characterization of the phenotype, a new cohort of 86 patients. Am J Med Genet A 2023; 191: 445–458
97) Shimada S, Shimojima K, Okamoto N, et al: Microarray analysis of 50 patients reveals the critical chromosomal regions responsible for 1p36 deletion syndrome-related complications. Brain Dev 2015; 37: 515–526
98) Arndt AK, Schafer S, Drenckhahn JD, et al: Fine mapping of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am J Hum Genet 2013; 93: 67–77
99) Jones KL, Jones MC, Campo MD: Deletion 4p syndrome, in Jones KL, Jones MC, Campo MD (eds): Smith’s Recognizable Patterns of Human Malformation, 8th eds, Philadelphia, Elsevier, 2022, pp32–33
100) Battaglia A, Carey JC, South ST: Wolf-Hirschhorn syndrome: A review and update. Am J Med Genet C Semin Med Genet 2015; 169: 216–223
101) Zollino M, Lecce R, Fischetto R, et al: Mapping the Wolf-Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region. Am J Hum Genet 2003; 72: 590–597
102) Shimizu K, Wakui K, Kosho T, et al: Microarray and FISH-based genotype–phenotype analysis of 22 Japanese patients with Wolf–Hirschhorn syndrome. Am J Med Genet A 2014; 164A: 597–609
103) Jones KL, Jones MC, Campo MD: Deletion 5p syndrome, in Jones KL, Jones MC, Campo MD (eds): Smith’s Recognizable Patterns of Human Malformation, 8th eds, Philadelphia, Elsevier, 2022, pp36–39
104) Nguyen JM, Qualmann KJ, Okashah R, et al: 5p deletions: Current knowledge and future directions. Am J Med Genet C Semin Med Genet 2015; 169: 224–238
105) Kondoh T, Shimokawa O, Harada N, et al: Genotype-phenotype correlation of 5p-syndrome: pitfall of diagnosis. J Hum Genet 2005; 50: 26–29
106) Jones KL, Jones MC, Campo MD: Smith-Magenis syndrome, in Jones KL, Jones MC, Campo MD (eds): Smith’s Recognizable Patterns of Human Malformation, 8th eds, Philadelphia, Elsevier, 2022, pp266–269
107) Falco M, Amabile S, Acquaviva F: RAI1 gene mutations: Mechanisms of Smith-Magenis syndrome. Appl Clin Genet 2017; 10: 85–94
108) Rinaldi B, Villa R, Sironi A, et al: Smith-Magenis syndrome: Clinical review, biological background and related disorders. Genes (Basel) 2022; 13: 335
109) Adam MP, Feldman J, Mirzaa GM, et al: Smith-Magenis syndrome. GeneReviews®. https://www.ncbi.nlm.nih.gov/books/NBK1310/
110) Boudreau EA, Johnson KP, Jackman AR, et al: Review of disrupted sleep patterns in Smith–Magenis syndrome and normal melatonin secretion in a patient with an atypical interstitial 17p11.2 deletion. Am J Med Genet A 2009; 149A: 1382–1391
111) Onesimo R, Versacci P, Delogu AB, et al: Smith–Magenis syndrome: Report of morphological and new functional cardiac findings with review of the literature. Am J Med Genet A 2021; 185: 2003–2011


This page was created on 2024-04-16T17:22:05.355+09:00
This page was last modified on 2024-05-27T17:27:34.000+09:00
このサイトは(株)国際文献社によって運用されています。