Noonan・RAS/MAPK症候群の小児循環器学BasicNoonan and RAS/MAPK Syndromes in Pediatric Cardiology: A Review
東京都立小児総合医療センターTokyo Metropolitan Children’s Medical Center ◇ Tokyo, Japan
発行日:2024年11月30日Published: November 30, 2024
Noonan症候群(Noonan syndrome: NS)は,特徴的顔貌,心疾患,低身長を特徴とする常染色体顕性(優性)遺伝形式の先天性異常症候群で,1963年にJacqueline Noonan博士によって報告された.多彩な全身症状に対して,包括的な診療が必要となる.近年,NSと臨床的に類似点が多いCostello 症候群(Costello syndrome: CS),心臓・顔・皮膚(cardio-facio-cutaneous: CFC)症候群などが,遺伝子解析によりNSと共通のRAS/MAPK細胞内シグナル伝達系の分子の異常により発症することが解明され,これらの症候群を含むNSの類縁疾患がRAS/MAPK症候群または“RASopathies”と総称されるようになった.本稿では,小児循環器専門医が知っておくべきNoonan・RAS/MAPK症候群の基本知識を紹介する.
Noonan syndrome (NS), first described by Dr. Jacqueline Noonan in 1963, is an autosomal dominant congenital anomaly syndrome. It is characterized by unique facial features, heart disease, and short stature. Patients with NS require comprehensive medical treatment for a variety of systemic symptoms. Genetic analyses have recently revealed that Costello syndrome and cardiofaciocutaneous syndrome, which share many clinical similarities with NS, are caused by abnormalities in the RAS/MAPK intracellular signaling pathway. Collectively, these syndromes and other NS-associated disorders are now referred to as RAS/MAPK syndromes or RASopathies. This review provides basic knowledge on 25 NS-associated disorders and RAS/MAPK syndromes with which pediatric cardiologists should be familiar.
Key words: Costello syndrome; cardiofaciocutaneous syndrome; PTPN11; RAF1; heart disease
© 2024 特定非営利活動法人日本小児循環器学会© 2024 Japanese Society of Pediatric Cardiology and Cardiac Surgery
Noonan症候群(Noonan syndrome: NS)は,特徴的顔貌,心疾患,低身長を特徴とする常染色体顕性(優性)遺伝形式の先天性異常症候群で,1963年にJacqueline Noonan博士によって報告された1).Fig. 1に示すような多彩な全身症状に対して,包括的な診療が必要となる2).近年,NSと臨床的に類似点が多いCostello症候群(Costello syndrome: CS),心臓・顔・皮膚(cardio-facio-cutaneous: CFC)症候群などが,遺伝子解析によりNSと共通のRAS/MAPK細胞内シグナル伝達系の分子の異常により発症することが解明され,これらの症候群を含むNSの類縁疾患がRAS/MAPK症候群または“RASopathies”と総称されるようになった3, 4).本稿では,小児循環器専門医が知っておくべきNoonan・RAS/MAPK症候群の基本知識を紹介する.
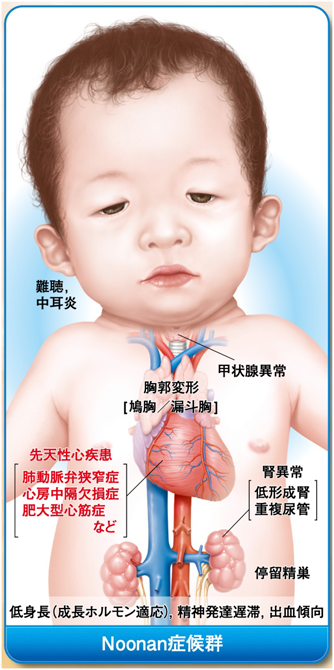
Noonan症候群では,図に示すような臨床徴候に対して,包括的診療を必要とする.文献2)より引用.
NSの約80%に心疾患が合併し,先天性心疾患の1.4%を占めると言われている.また,心疾患を高率に合併する症候群として21trisomyに次いで2番目に多い5).
NSの診断は基本的に臨床診断であり,心疾患と特徴的顔貌により経験ある小児循環器専門医に初めて診断されることが多い.臨床診断にはTable 1に示す診断基準の主要徴候に注目し,副徴候を参考とする6, 7).診断基準(Table 1)は,2023年10月に日本医療研究開発機構難治性疾患実用化研究事業「ヌーナン症候群類縁疾患の診断・診療ガイドライン作成に向けたエビデンス創出研究」(研究開発代表者:青木洋子)により,アップデートされた.臨床診断によることを原則としたうえで,可能であれば遺伝子診断を実施することが望ましい.遺伝子異常が同定されるのは60~80%で,全例ではないことに留意する.遺伝子検索の結果,PTPN11,SOS1,RAF1,NRAS,SOS2,RIT1,KRAS,LZTR1のいずれかにNSの病因と考えられる遺伝子異常が検出された場合,確定診断としてよい7, 8).検査は保険収載され,かずさDNA研究所で検査できる.
 |
| (文献7)よりTable 1を引用) |
心疾患,特に小児で肺動脈弁狭窄症(pulmonic stenosis: PS),肥大型心筋症(hypertrophic cardiomyopathy: HCM)を診断した場合,Table 1に示されたNSの身体的特徴の有無を念頭に鑑別診断することも重要である.Table 1の留意点として,(A-2)心臓の表現型として以前より閉塞性肥大型心筋症(hypertrophic obstructive cardiomyopathy: HOCM)と記載されているが,実際のNSのHCMは必ずしも閉塞性ではない(Table 1(A-2)のHOCMの表記については,2024年現在ガイドライン委員会で検討中である).NSは常染色体顕性(優性)遺伝形式をとるため,NSと診断された児の親もNSであることが疑われる場合もある.親の心疾患についての精査も勧められるが,それまで健常と考えられてきた親の診断においては,本人の心情や家族関係に十分配慮した遺伝カウンセリングが必要である.
RAS/MAPK症候群には,神経線維腫症I型(neurofibromatosis I: NF1),NS,LEOPARD症候群と呼ばれていたNoonan syndrome with multiple lentigines(NSML),capillary malformation–arteriovenous malformation Syndrome(CM-AVM),CS,CFC症候群,Legius症候群が含まれる.これらの疾患・症候群は,生命維持に重要な役割を果たすRAS/MAPK細胞内シグナル伝達系の分子をコードする遺伝子の異常を病因とする.Fig. 2およびTable 2に示すように,共通の分子経路の中の異なる分子(遺伝子)の異常により,それぞれの疾患・症候群が発症する3, 4).
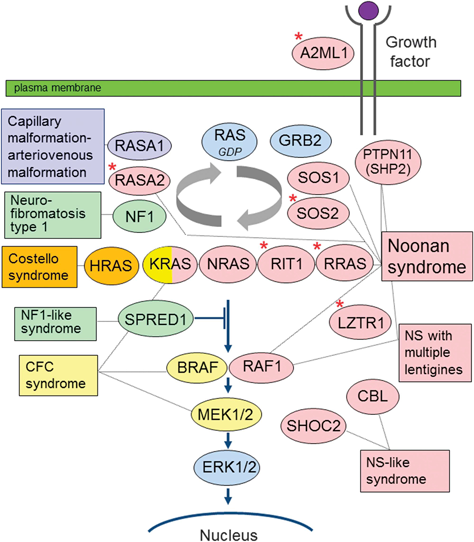
Noonan・RAS/MAPK症候群は,生命維持に重要な役割を果たすRAS/MAPK細胞内シグナル伝達系の様々な分子をコードする遺伝子の異常を病因とする.文献3)より引用.
| 遺伝子変異(頻度) | 特徴的な心疾患 |
|---|---|
| PTPN11(~50%) | 肺動脈弁狭窄症,心房中隔欠損症 |
| 肥大型心筋症は少ない | |
| SOS1(11%) | 肺動脈狭窄 |
| RAF1(5%) | 肥大型心筋症 |
| SHOC2(~2%) | 僧帽弁逸脱と中隔欠損 |
| KRAS(~1.5%) | 肺動脈狭窄 |
| RIT1 (5%) | 肥大型心筋症 |
| MEK1 (?%) | 肺動脈狭窄 |
| ( )内の数字はNoonan症候群における頻度を示す.特に特徴的な心疾患が報告されていない遺伝子変異NRAS(0.2%), CBL, RRAS, RASA2, A2ML1, SOS2, LZTR1, BRAF | |
日本小児循環器学会・遺伝子疫学委員会の調査(平成23–25年度研究課題)では,遺伝子検査が実施されたNoonan類縁疾患82例のうち,遺伝子異常が判明したのは48例(59%)であった9).内訳は,PTPN11変異が30例(63%)で最も多かった.PTPN11変異例にはPSが多かったが,遺伝子変異と治療の必要性には関連性はなかった.心房中隔欠損症(atrial septal defect: ASD)もPTPN11変異例で多く,約6割で手術が行われていた.HCMはRAF1変異6例全例(100%)に認められ,全例で内服治療が行われていた一方,PTPN11変異例では7例(23%)で,うち3例(43%)だけが内服治療を受けていた.また,NSMLの3例中2例でPTPN11変異があり,HCMとPSを合併していた.MEK2変異の1例では,僧帽弁逸脱・逆流が認められた.CFC症候群の3例中,KRAS変異が1例,BRAF変異が1例に検出され,PS 1例,HCM 1例,HCMとPSの合併1例だった.CS 3例中2例にHRAS変異が認められ,HCMとPSが1例ずつだった.CSのもう1例にはSHOC2変異があり,僧帽弁閉鎖不全術後に不整脈で突然死していた.
NSの診断基準・主要徴候の1項目に,「特徴的な心電図所見」がある(Table 1).この「特徴的な心電図所見」はNS症例の約60%に認められると報告されており,Fig. 3に示すようなaVF誘導のQRS電気軸陰性を伴う左軸偏位,左側前胸部誘導のR/S比の異常,異常Q波で,通常右側前胸部誘導で見られるrSパターンが左側前胸部誘導でも連続して見られる,いわゆる“clockwise rotation”の所見である10).この所見は,心疾患の有無と関連なく認められ,心電図所見に精通した小児循環器医がNSを診断する際に非常に役立つ.
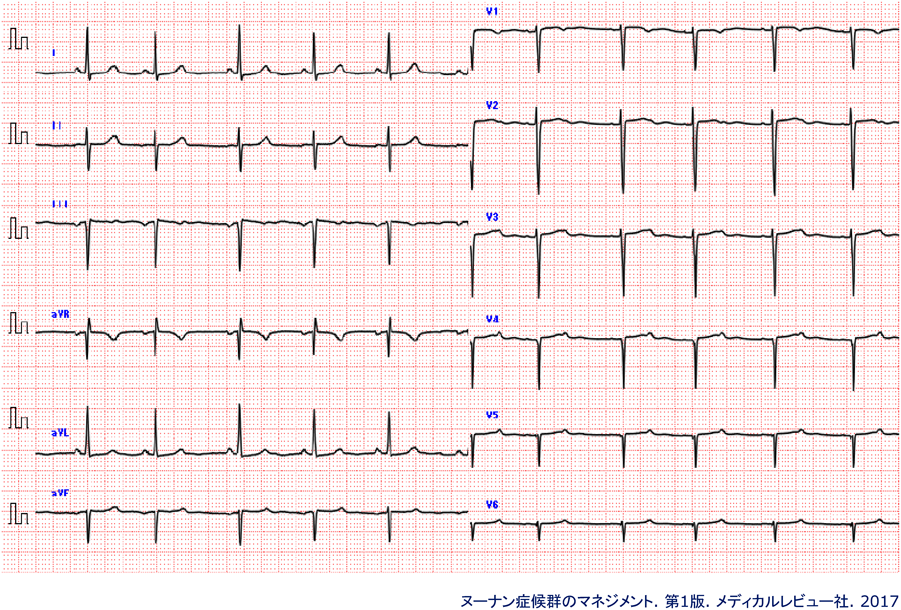
詳細は本文を参照.
PSが20~50%と最も多く,PSの7%をNSが占めるといわれる.さらに二次孔型ASDの合併も高率(10~20%)で11),不完全型房室中隔欠損症(一次孔型ASD)もスペクトラムに含まれる12).NSでは右心系の異常が多く認められるのが特徴であり,左心系閉塞性心疾患(大動脈弁狭窄症,大動脈縮窄症など)を特徴とするTurner症候群と対比される.また,HCMが20~30%に合併し,心室拡張機能障害(拘束型心筋症様)を呈するのも特徴である13).その他,Fallot四徴症,大動脈縮窄症,僧帽弁異常を認める例もあるが,不整脈や突然死の発生は少ないと言われている.
NS以外のRAS/MAPK症候群の心血管疾患の特徴をTable 3にまとめる3, 4).小児における多源性心房頻拍(multifocal atrial tachycardia: MAT)は通常特発性だが,肺疾患や先天性心疾患の術後,またCSに合併することが報告されている.この機序は不明だが,重度なPSやHCMと関連なく,独立した不整脈として存在する.
| 疾患 | 心疾患 |
|---|---|
| NS with multiple lentigines(LEOPARD症候群) | 肥大型心筋症(80%に心構造異常),心電図異常(73%),弁異常(50%),肺動脈狭窄(23%),冠動脈異常(15%) |
| NS-like disorder with loose anagen haer | 肺動脈弁狭窄(39%),僧帽弁異形成(31%),肥大型心筋症(27%),中隔欠損症(42%) |
| NS-like disorder | 心筋症,不整脈,大動脈弁や僧房弁異常 |
| Costello症候群 | 肥大型心筋症(60%),先天性心疾患(44%),肺動脈弁狭窄(22%),心房頻脈(48%) |
| CFC(Cardiofaciocutaneous)症候群 | 肺動脈弁狭窄(45%),肥大型心筋症(40%),中隔欠損症,弁異常,不整脈,大動脈拡張 |
| Nerofibromatosis type1 (NF1) | NF1 vasculopathy(瘤,狭窄,動静脈異常),高血圧,肺動脈弁狭窄,先天性心疾患,心内神経線維腫症 |
| NF1 like syndrome(Legius syndrome) | 肺動脈弁狭窄,僧帽弁逸脱 |
NSでは,肺動脈弁異形成によるPSが特徴的である.25~35%の症例で肺動脈弁が異形成で,しばしばASDを合併する14–16).異形成弁は3弁が多く,著明に厚い弁尖は粘液腫様の組織で構成され,癒合が見られる.弁輪は通常低形成である.重度の肺動脈弁異形成は,症候群でないPSではあまり見られないため,NSを疑う心臓所見である.
自然歴として,軽度のPSは平均13.6年の経過観察では進行しなかった,とする報告がある.また,NSのPSの58%は無治療経過観察で,約1/3は開胸手術もしくは複数回の治療を必要とした,とする報告もある8).PSに対する治療の第一選択であるバルーン拡張術は禁忌ではないが,異形成弁に対して狭窄の解除効果に乏しく,肺動脈損傷による出血の危険があるので注意を要する.
心臓の弁は,房室管と流出路に形成される心内膜床(cardiac cushions)の部分に発生する.心臓の原基である(原始)心筒は,内側の内皮(心内膜)細胞層と外側の心筋層と,それらを分ける細胞間基質によって構成されている.心筒の屈曲(ルーピング)と同時に,心室および心房形成部分では心筋層が発達するが,房室管と流出路の部分では心筋層は発達しない.房室管では,内皮細胞が形質転換して細胞間基質の層に流入し,間葉系細胞に分化・増殖して心内膜床を形成していく.流出路では,神経管背側から移動して細胞間基質の層に流入する心臓神経堤細胞から分化した間葉系細胞の関与が大きい.したがって,僧帽弁と三尖弁がもともと局所の内皮細胞に由来する心内膜床の組織だけで形成されるのに対し,大動脈弁と肺動脈弁の発生には心臓神経堤細胞も関与すると考えられている8, 14–16).
正常の弁は,心筒内腔に膨隆した心内膜床として形成された組織がリモデリングすることにより,一層の内皮細胞と中心基質にコラーゲン・エラスチン・グリコサミノグリカンを有する薄い葉状構造物として形成される.したがってNSで見られる異形成弁は,このリモデリングの過程が起こらず,分厚く可動性の悪い構造になった結果と推測される.NSのモデル動物PTPN11変異マウスでは,間葉系細胞の異常増殖による半月弁の異常が認められ,RAS/MAPKシグナル経路の恒常的な亢進によって,心内膜床形成からリモデリングへの変換が障害される機序が示唆される16).ただし,このマウスでは肺動脈弁だけでなく,大動脈弁にも同様の異常が認められることが,ヒトNSと異なる.心内膜床の間葉系細胞に発現するGタンパク結合受容体CXCR7を欠失するマウス胎仔では,BMPシグナルが増加し,間葉系の弁の細胞が過剰に増殖することにより大動脈弁と肺動脈弁の狭窄を起こす.モデル動物を用いた研究により,半月弁の発生とその異常について解明が進んでいるが,それぞれの半月弁の形成を特異的に制御する分子機構は不明で,なぜNSでは肺動脈弁が特に障害されるのかはわかっていない.
HCMはNSの約20%で見られる.遺伝子型では,RAF1変異で頻度が高く,重症例も多い.NSのPTPN11変異ではHCMの頻度が少ない一方,NSMLのPTPN11変異ではHCMの合併が多いことは興味深い(後述).KRAS変異では重症HCMが多く,SHOC2変異では僧帽弁逸脱と中隔欠損の合併が見られるが,NS全体での重症度と自然歴は様々である14).特発性HCMと比較すると,NSでは乳児期の発症が57%で,より早期に発症する.乳児期にHCMと診断された児の死亡率は15%と高く,先天性心疾患を合併する頻度も70%と高い.乳児期以降の発症例については,成人期まで安定している症例,改善する症例がある一方,進行する症例もあり,様々な経過が報告されている.年次死亡率は非症候群性のHCMとほぼ同じで,突然死や不整脈の報告も多くない13–15, 17–19).
HCMはしばしばサルコメア病とされ,心筋収縮の分子的な力を生み出しているサルコメアタンパクをコードする遺伝子のほぼすべてで病因変異が同定されている.主な遺伝子は,MYBPC3とMYH7で,検出される変異の約70%を占める20).NSの病因遺伝子であるPTPN11にコードされるSH2 domain-containing protein tyrosine phosphatase(SHP2)は,サルコメアタンパク以外でHCMに関連するシグナルタンパクであり,RASopathyに関連する心筋症はサルコメア病と異なる「二次性心筋症」に分類される.PTPN11変異はNSで約50%,NSMLではほとんどの症例で検出され,NSMLで主としてHCMに関連する一方,NSではむしろPS,ASDとの関連が多く,HCMは少ない.これまで基礎研究でNSのPTPN11変異は機能獲得変異(gain of function)であり,SHP2のphosphatase活性を増強する一方,NSMLのPTPN11変異は機能喪失変異(loss of function)であり,PTPの触媒作用に重要な保存残基に影響しphosphatase活性を減弱することが報告されている.PI3K/AKT経路の調節不全がNSMLにおける心臓の病態生理に関与し,SHP2Y279CとSHP2T468M変異がGAB1(Grb2-associated binding protein1)の脱リン酸化を阻害し,PI3K/AKT活性を増加させることにより心筋細胞の肥大を促進する結果が報告されている21).また,NSでHCMを高率に認める遺伝子変異として,RAF1およびRIT1変異が報告されている22).RAF1変異のHCMおいてはMEK1/2-ERK1/2mitogen-activated protein kinasesの活性化やERK effector type 3 p90 ribosomal S6 kinase(RSK3),calcineurin-NFAT系の関与が報告されている22–24).RIT1変異におけるHCMの機序に関しての報告はまだないが,遺伝子変異を導入したゼブラフィッシュにおいてERK1/2が活性化されることが報告されている25).
NSは常染色体顕性(優性)遺伝の疾患だが,常染色体潜性(劣性)遺伝のNS家系においてleucine zipper like transcription regulator 1(LZTR1)遺伝子の異常が病因になることが,日本の研究室から明らかにされた26, 27).LTZR1は染色体22q11.21に座位し,BTB-Kelchスーパーファミリーに属するがん抑制遺伝子で,細胞内ではGolgiへの局在が示唆されている.RAS/MAPKシグナルとの関連として,RASタンパクのユビキチン化によるdegradationを促進することが示唆されているが,RAS/MAPKシグナル伝達分子そのものではない,Noonan・RAS/MAPK症候群の病因としてはユニークな分子である.
近年の分子標的治療薬の進歩により,Noonan・RAS/MAPK症候群に対してもFig. 4に示すようなシグナル伝達経路阻害薬の治療応用が理論的に考えられる28).症例報告レベルだが,これまでに乳児NSMLのHCMに対してmTOR阻害薬everolimus29),RIT1変異陽性NS30, 31)およびRAF1変異陽性NS32)のHCMに対してMEK阻害薬trametinib,SOS1変異陽性NSのリンパ管異形成33)およびRIT1変異陽性NSの乳び胸34)に対してMEK阻害薬trametinibなどの治療経験が報告されており,今後さらにトライアルが進展することが期待される.
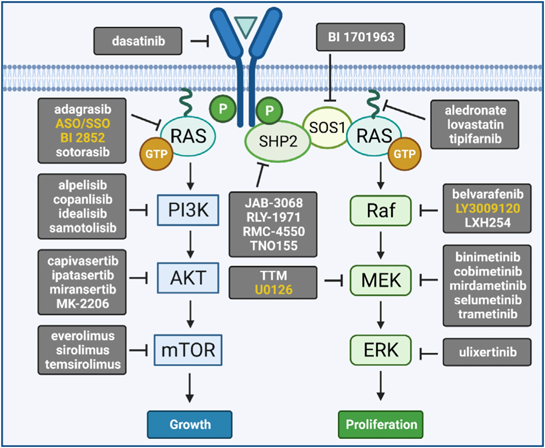
RAS/MAPKシグナル伝達系の各分子を標的とした抑制薬を示す.詳細は本文を参照.文献28)より図を改訂.
NSの低身長に対して35),日本ではGH治療が2017年に承認された.実施については小児内分泌専門医と連携することが望ましく,注意点は以下のとおり:
①若年性骨髄単球性白血病(JMML)との関連について
PTPN11遺伝子変異を代表とするRAS-MAPK経路の機能獲得型遺伝子変異は,悪性血液疾患,とくに若年性骨髄単球性白血病(juvenile myelomonocytic leukemia: JMML)を引き起こす原因と考えられている.Noonan症候群にJMMLの合併が報告されているが,多くが良性・一過性である.一方,JMMLの単発例は侵襲的であり悪性度が高いことが知られており,しばしばより強力な機能獲得変異を有する.強力な機能獲得変異では,Noonan症候群と診断されずに新生児期に致死的経過をたどる症例もあると推測されている36, 37).
②固形腫瘍の発症リスクについて
胎児性腫瘍の発症リスクも通常より高いと考えられている.上腹部超音波検査などにより経過観察を行う.
③GH投与による心筋症の発症・増悪の可能性について
現在までにGH投与が心筋症の発症・増悪に関連するというデータは存在しないが,その可能性が懸念されている.GH治療中,心臓超音波検査などにより経過観察を行う(GH治験時同様,半年に1回程度の検査が望ましい).
我が国における臨床試験開始から8年間70例(臨床試験4年間から継続例35例,市販後調査新規例35例)の報告がまとめられ,NSに対するGH治療の有効性と安全性が確認された.ただし,RAF1変異を有するNSの男児1例の死亡(死亡時年齢12歳)が報告されている38).死因は閉塞性HCMに起因する致死性不整脈で,臨床試験当初からHCMと不整脈に対する治療中で,GH投与中約7年間でHCMの悪化がなかったことから,報告医師からはGH治療との関連は“unlikely”とされ,製薬会社からは“impossible to assess”とされている.GH治療によるHCM悪化のエビデンスはないが,心臓超音波検査などにより注意深く経過観察することが必要と考えられる.
GH治療を念頭においた「NSと小児循環器医の役割」の動画(約5分)が,日本小児循環器学会ホームページ(【会員専用】ページ)で公開されている.ご興味のある方には,ぜひご視聴いただきたい.
〈ホームページ〉https://jspccs.jp/
HOME >各種活動・報告>研究委員会>課題研究委員会活動報告【会員専用】
NSに対しては,移行医療を含めて生涯にわたり心臓の経過観察が必要である39).特にHCMの症例では,思春期以降に発症する例もあり,左室流出路閉塞性病変が成人期に進行するおそれがある.PSに対して治療した症例では,遠隔期の肺動脈弁の機能不全,右室機能不全に留意する必要がある.成人期の長期フォローアップの報告は限られているが,不整脈は少ないとされている8).
本稿について,開示すべき利益相反はない.
1) Noonan JA, Ehmke DA: Associated non cardiac malformations in children with congenital heart disease. J Pediatr 1963; 63: 468–470
2) 山岸敬幸:顔貌でわかる先天性心疾患.https://publish.m-review.co.jp/files/tachiyomi_J0055_1101_0005-0006.pdf
3) Aoki Y, Niihori T, Inoue S, et al: Recent advances in RASopathies. J Hum Genet 2016; 61: 33–39
4) Jhang WK, Choi JH, Lee BH, et al: Cardiac manifestations and associations with gene mutations in patients diagnosed with RASopathies. Pediatr Cardiol 2016; 37: 1539–1547
5) Marino B, Digilio MC, Toscano A, et al: Congenital heart diseases in children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr 1999; 135: 703–706
6) van der Burgt I, Berends E, Lommen E, et al: Clinical and molecular studies in a large Dutch family with Noonan syndrome. Am J Med Genet 1994; 53: 187–191
7) 青木洋子,ほか:ヌーナン症候群の診断基準.http://jspe.umin.jp/medical/files/guide20231003.pdf
8) Shaw AC, Kalidas K, Crosby AH, et al: The natural history of Noonan syndrome: A long-term follow-up study. Arch Dis Child 2007; 92: 128–132
9) 日本小児循環器学会・心血管疾患の遺伝子疫学委員会,平成23-25年度研究課題(短期目標)報告書): Noonan症候群update. https://jspccs.jp/member/report/studycommittee/index.php?download=noonan2019.pdf
10) Raaijmakers R, Noordam C, Noonan JA, et al: Are ECG abnormalities in Noonan syndrome characteristic for the syndrome? Eur J Pediatr 2008; 167: 1363–1367
11) Sharland M, Burch M, McKenna WM, et al: A clinical study of Noonan syndrome. Arch Dis Child 1992; 67: 178–183
12) Marino B, Digilio MC, Toscano A, et al: Congenital heart diseases in children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr 1999; 135: 703–706
13) Burch M, Sharland M, Shinebourne E, et al: Cardiologic abnormalities in Noonan syndrome: Phenotypic diagnosis and echocardiographic assessment of 118 patients. J Am Coll Cardiol 1993; 22: 1189–1192
14) Jhang WK, Choi JH, Lee BH, et al: Cardiac manifestations and associations with gene mutations in patients diagnosed with RASopathies. Pediatr Cardiol 2016; 37: 1539–1547
15) Katherine A: The RASopathies. Annu Rev Genomics Hum Genet 2013; 14: 355–369
16) Chen B, Bronson RT, Klaman LD, et al: Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nat Genet 2000; 24: 296–299
17) Prendiville TW, Gauvreau K, Tworog-Dube E, et al: Cardiovascular disease in Noonan syndrome. Arch Dis Child 2014; 99: 629–634
18) Colquitt JL, Noonan JA: Cardiac findings in Noonan syndrome on long-term follow-up. Congenit Heart Dis 2014; 9: 144–150
19) Romano AA, Allanson JE, Dahlgren J, et al: Noonan syndrome: Clinical features, diagnosis, and management guidelines. Pediatrics 2010; 126: 746–759
20) Buikema JW, Wu SM: Untangling the biology of genetic cardiomyopathies with pluripotent stem cell disease models. Curr Cardiol Rep 2017; 19: 30
21) Lauriol J, Cabrera JR, Roy A, et al: Developmental SHP2 dysfunction underlies cardiac hypertrophy in Noonan syndrome with multiple lentigines. J Clin Invest 2016; 126: 2989–3005
22) Aoki Y, Niihori T, Inoue S, et al: Recent advances in RASopathies. J Hum Genet 2016; 61: 33–39
23) Passariello CL, Martinez EC, Thakur H, et al: RSK3 is required for concentric myocyte hypertrophy in an activated Raf1 model for Noonan syndrome. J Mol Cell Cardiol 2016; 93: 98–105
24) Dhandapany PS, Fabris F, Tonk R, et al: Cyclosporine attenuates cardiomyocyte hypertrophy induced by RAF1 mutants in Noonan and LEOPARD syndromes. J Mol Cell Cardiol 2011; 51: 4–15
25) Koenighofer M, Hung CY, McCauley JL, et al: Mutations in RIT1 cause Noonan syndrome: Additional functional evidence and expanding the clinical phenotype. Clin Genet 2016; 89: 359–366
26) Abe T, Umeki I, Kanno S, et al: LZTR1 facilitates polyubiquitination and degradation of RAS-GTPases. Cell Death Differ 2020; 27: 1023–1035
27) Nakagama Y, Takeda N, Ogawa S, et al: Noonan syndrome-associated biallelic LZTR1 mutations cause cardiac hypertrophy and vascular malformations in zebrafish. Mol Genet Genomic Med 2020; 8: e1107
28) Gelb BD, Yohe ME, Wolf C, et al: New prospectives on treatment opportunities in RASopathies. Am J Med Genet C Semin Med Genet 2022; 190: 541–560
29) Hahn A, Lauriol J, Thul J, et al: Rapidly progressive hypertrophic cardiomyopathy in an infant with Noonan syndrome with multiple lentigines: Palliative treatment with a rapamycin analog. Am J Med Genet A 2015; 167A: 744–751
30) Andelfinger G, Marquis C, Raboisson M-J, et al: Hypertrophic cardiomyopathy in Noonan syndrome treated by MEK-inhibition. J Am Coll Cardiol 2019; 73: 2237–2239
31) Leegaard A, Gregersen PA, Nielsen TØ, et al: Successful MEK-inhibition of severe hypertrophic cardiomyopathy in RIT1-related Noonan Syndrome. Eur J Med Genet 2022; 65: 104630
32) Mussa A, Carli D, Giorgio E, et al: MEK inhibition in a newborn with RAF1-associated Noonan syndrome ameliorates hypertrophic cardiomyopathy but is insufficient to revert pulmonary vascular disease. Genes (Basel) 2022; 13: 6
33) Dori Y, Smith C, Pinto E, et al: Severe lymphatic disorder resolved with MEK inhibition in a patient with Noonan syndrome and SOS1 mutation. Pediatrics 2020; 146: e20200167
34) Nakano TA, Rankin AW, Annam A, et al: Trametinib for refractory chylous effusions and systemic complications in children with Noonan syndrome. J Pediatr 2022; 248: 81–88
35) Chacko EM, Rapaport R: Short stature and its treatment in Turner and Noonan syndromes. Curr Opin Endocrinol Diabetes Obes 2012; 19: 40–46
36) Strullu M, Caye A, Lachenaud J, et al: Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet 2014; 51: 689–697
37) Mason-Suares H, Toledo D, Gekas J, et al: Juvenile myelomonocytic leukemiaassociated variants are associated with neo-natal lethal Noonan syndrome. Eur J Hum Genet 2017; 25: 509–511
38) Muroya K, Kawai M, Yamagishi H: Growth hormone effectively improved height outcomes with no new safety signals in Japanese children with Noonan syndrome. In submission
39) 日本循環器学会/日本心臓病学会/日本小児循環器学会合同ガイドライン:2024年改訂版 心臓血管疾患における遺伝学的検査と遺伝カウンセリングに関するガイドライン.https://www.j-circ.or.jp/cms/wp-content/uploads/2024/03/JCS2024_Imai.pdf


This page was created on 2024-12-03T11:18:38.226+09:00
This page was last modified on 2025-03-11T09:46:32.000+09:00
このサイトは(株)国際文献社によって運用されています。