二次性心筋症の診断Diagnosis of Secondary Cardiomyopathies
北海道大学大学院医学研究院 小児科学教室Department of Pediatrics, Hokkaido University Graduate School of Medicine ◇ Hokkaido, Japan
発行日:2024年11月30日Published: November 30, 2024
二次性心筋症は鑑別疾患が重要であり,小児では代謝性疾患や神経筋疾患に合併することが多い.発症年齢は原因によっても異なり,鑑別において重要である.本総説では小児から成人移行期に発症する代表的な二次性心筋症であるFabry病,Pompe病,Danon病,ミトコンドリア心筋症,RAS/MAPK症候群,Duchenne/Becker型筋ジストロフィー症合併心筋症の発症機序および診断法について,および二次性心筋症の鑑別における心筋生検の役割について解説する.
In cases of secondary cardiomyopathies, it is crucial to distinguish between background diseases, such as metabolic and neuromuscular diseases, that are often seen in children. The age at onset varies according to the cause and is important for the differential diagnosis. This review discusses the diagnosis and pathogenesis of cardiomyopathy associated with Fabry disease, Pompe disease, Danon disease, mitochondrial disease, RAS/MAPK syndrome (RASopathies), and Duchenne/Becker muscular dystrophy, which often occur in children and adults in transition. We also discuss the role of myocardial biopsy in the identification of secondary cardiomyopathies.
Key words: secondary cardiomyopathy; hypertrophic cardiomyopathy; endomyocardial biopsy; genetic testing; electron microscopy
© 2024 特定非営利活動法人日本小児循環器学会© 2024 Japanese Society of Pediatric Cardiology and Cardiac Surgery
二次性心筋症の概念については学会によっても異なるため注意が必要である.2006年に提唱された米国心臓病学会(ACC)および米国心臓協会(AHA)のガイドラインでは,心筋病変が主座ではなく全身疾患の一部である場合に二次性心筋症と定義し,代謝性疾患,神経筋疾患,膠原病,放射線/化学療法後,RAS/MAPK症候群に伴う心筋障害などがそれにあたる1).2008年に提唱された欧州心臓病学会(ESC)の分類では二次性心筋症という概念が排除された分類となっている2).しかしながら,実臨床で遭遇する肥大型心筋症,拡張型心筋症などに際しては二次性心筋症を除外診断する必要があり,2018年に提唱された日本循環器学会のガイドラインは二次性心筋症を可能な限り鑑別することを重視した分類となっている3).本稿ではそのなかでも特に小児期~成人移行期に発症する頻度が高い二次性心筋症について概説する.
二次性心筋症の中には心筋肥大を呈してくるものがあり,肥大型心筋症様心肥大と呼ばれているが,その鑑別診断において発症年齢は極めて重要である.例えば,心アミロイドーシスやFabry病は小児期に発症することは稀であるが,Pompe病,RAS/MAPK症候群に伴う肥大型心筋症様心肥大は新生児~乳児期に発症する.また,ミトコンドリア心筋症のように初発年齢がすべての年齢層にまたがる場合もあり,発症年齢による疾患頻度を考慮した鑑別診断が必要である.鑑別すべき代表的疾患の臨床的特徴を表に示す(Table 1).また,発症年齢と疾患頻度で表した小児二次性心筋症のイメージ図を示す(Fig. 1)4).
| Onset of cardiomyopathy | Neonate–Infant | Neonate–Infant | Neonate–Adult | Childhood–Adolescent |
|---|---|---|---|---|
| Primary disease | RASopathies | Pompe disease (Infantile-onset) | Mitochondrial disease | Propionic acidemia |
| Complication rate of cardiomyopathy | 10–29% | Almost (<2 years old) | 17–40% | unknown |
| Cardiac phenotype | HCM | HCM | HCM, d-HCM, DCM, RCM, LVNC | DCM |
| Genetic form | AD | AR | All forms of inheritance including maternal inheritance | AR |
| Causative gene | PTPN11, RAF1, RIT1 | GAA | Over 300 genes including mitochondrial DNA | PCCA, PCCB |
| Treatment | beta-blockers, cibenzoline, myectomy (for HOCM) | ERT | vitamin cocktail (for mitochondrial crisis) | low-protein diet, L-carnitine, metronidazole, liver transplantation |
| Prognosis | SR 30% at 2 years old (early infantile-onset) | SR 25% at 1 year old without ERT | Cumulative SR <20% at 150 months | Very poor (early infantile-onset) unknown (late onset) |
| Onset of cardiomyopathy | Childhood–Adolescent | Adolescent–Adult | Adolescent–Adult | Adult |
| Primary disease | Propionic acidemia | Danon disease | DMD/BMD | Fabry disease |
| Complication rate of cardiomyopathy | unknown | 11–12% | 60–75% | 50% |
| Cardiac phenotype | DCM | HCM, d-HCM | DCM | HCM |
| Genetic form | AR | XD | XR | X-linked |
| Causative gene | PCCA, PCCB | LAMP2 | DMD | GLA |
| Treatment | low-protein diet, L-carnitine, metronidazole, liver transplantation | beta-blockers, ICD implantation, heart transplantation | supportive care, steroids, ACEi, beta-blockers | ERT, Pharmacologic Chaperone Therapy, Substrate Reduction Therapy |
| Prognosis | Very poor (early infantile-onset) unknown (late onset) | Affected males were unlikely to live until age 25 without heart transplantation | A median life expectancy of approximately 30 years | Median cumulative survival is approximately 50 years |
| ACEi, angiotensin converting enzyme inhibitor; AD, Autosomal dominant; AR, Autosomal recessive; d-HCM, dilated phase of hypertrophic cardiomyopathy; DMD/BMD, Duchenne muscular dystrophy/Becker muscular dystrophy; ERT, enzyme replacement therapy; HCM, hypertrophic cardiomyopathy; HOCM, hypertrophic obstructive cardiomyopathy; ICD, implantable cardioverter defibrillator; LVNC, left ventricular non-compaction; RCM, restrictive cardiomyopathy; SR, survival rate; XD, X-linked dominant; XR, X-linked recessive. | ||||
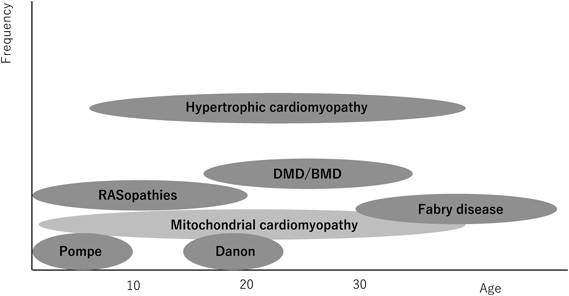
DMD/BMD, Duchenne muscular dystrophy/Becker muscular dystrophy. Modified from ref 4).
ライソゾーム病(先天代謝異常症)
X連鎖遺伝.男性のみに発症する疾患で女性は保因者となるとされてきたが,女性(ヘテロ接合体)でも症状が発現することが報告されている.
グロボトリアシルセラミド(Gb3)を分解する酵素αガラクトシダーゼA(α-galactosidase A: GLA)の遺伝子(GLA)の病的バリアントによってGb3が体内に蓄積するライソゾーム病である.蓄積分子別ではスフィンゴ脂質が蓄積する疾患の一つである.スフィンゴ脂質とはスフィンゴシンに脂肪酸がついたもので別名セラミドと呼ばれ保水性を有するため保湿剤に配合される化合物である.セラミドに糖鎖が3個ついたものがGb3である(Fig. 2).この最初の糖鎖を分解する酵素がGLAであり,この酵素欠損によって小児期からGb3が汗腺に蓄積して無汗症や体温調節障害などを来すほか,末梢神経障害による四肢末端痛を認める.腎障害は糸球体や尿細管にGb3が蓄積し,尿蛋白,尿細管障害を来す.小児期は軽症で見逃されやすいが,尿中マルベリー細胞はFabry病に特異的な所見であり,酵素補充療法のトリガーになるため重要である.成人以降では心筋にも蓄積し肥大型心筋症様心肥大を呈する.
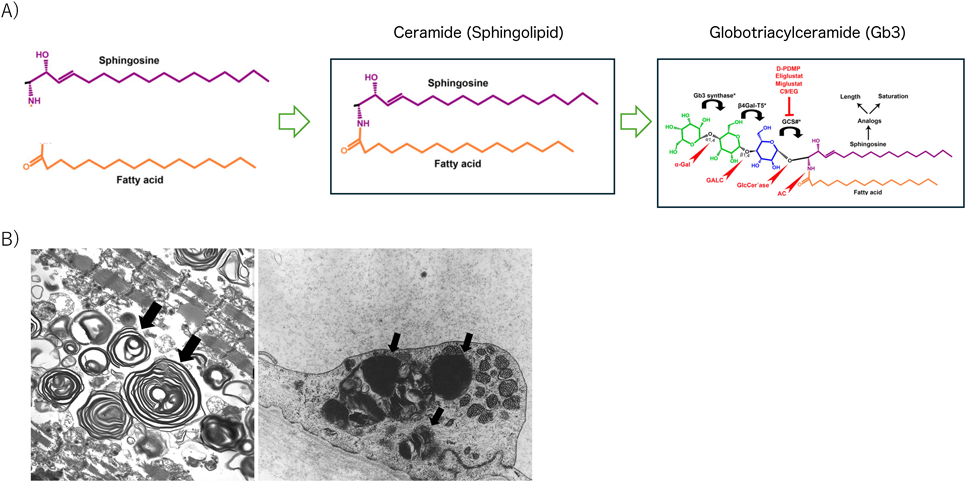
(A) Sphingosine bound to fatty acids is known as ceramide (sphingolipid). Ceramides with attached sugar chains are known as globosides. Ceramides with three attached sugar chains are called globotriaosylceramides (Gb3). (B) Electron microscopy showing typical intracellular storage of Gb3 forming “zebra bodies” within the myocardial cells (left) and endothelial cells (right) (black arrow). Modified from ref 5).
進行性の心肥大が特徴的である.男性では20歳代,女性では30歳代以降に全周性心肥大を生じ,進行例では左室後壁基部の壁運動異常と菲薄化がみられる.心電図ではストレインパターンを伴う左室高電位が肥大出現前に認められる.Fabry病では糖脂質が心筋に沈着するため心臓MRIにおけるT1値が低値となり,特異的所見として鑑別診断につながることがある.Gb3はセラミド特有の層状構造をもつため,心筋病理における電子顕微鏡像では同心円状層構造物(Zebra body)として本症に特異的な蓄積物質として確認され,鑑別診断上重要な所見である5).
小児期から学童期までの間に四肢疼痛,無汗症,被核血管腫などの症状が出現するが見逃されていることがある.成人以降では末期腎障害,脳血管病変として慢性白質病変,脳底動脈の拡張,微小出血病変が認められ,若年脳梗塞症例の一部にFabry病患者が含まれていることもある.
男性で白血球GLA活性が正常の約10%以下であれば確定診断となる.女性は酵素活性で診断することはできない.いずれの場合でも病的遺伝子バリアントが同定されれば確定診断となるが,未報告の遺伝子バリアントについては病原性の有無についての判断が必要となる.
ファブリー病遺伝子検査(かずさ遺伝子検査室),対象遺伝子:GLA,保険点数3,880点
酵素補充療法は2004年から保険収載されており,長期的な臓器障害および生命予後の改善効果が認められている.一方,点滴静注で2週ごとに反復投与が必要であることや,アナフィラキシーを含むアレルギー反応や抗体産生による効果減弱も報告されていることが欠点である.薬理学的シャペロン療法は2018年より保険収載されており,GLA変異酵素に結合して酵素活性を発揮する.経口薬であることが利点で心肥大抑制効果も高い6)が,遺伝子バリアントの部位によって適応が限定されることが欠点である.基質合成阻害療法はGb3を合成するグルコシルセラミド合成酵素を阻害することでGb3そのものを減らす治療法であり,現在治験が進められている.
ライソゾーム病(先天代謝異常症)
常染色体潜性遺伝(劣性遺伝)
グリコーゲンの分解に関わる酸性α-グルコシダーゼ(α-Glucosidase: GAA)の遺伝子(GAA)の病的バリアントにより酵素活性低下あるいは欠損をきたしてグリコーゲンが骨格筋,肝,心筋などに蓄積する.発症時期により,乳児型,小児型,成人型に分けられる.
乳児型では肥大型心筋症様心肥大を認め,自然経過では心不全または呼吸不全で2歳までに死亡する.小児型では2歳以降の発症で心肥大を来すことは稀である.
乳児型では,筋緊張低下,肥大型心筋症様心肥大,不整脈,運動発達遅滞,哺乳障害,肝腫大,巨舌,呼吸障害などを呈し,生後2か月から数か月以内で発症する7)(Fig. 3).小児型では生後6か月~幼児期より進行する筋力低下で発症し,20歳以降で呼吸不全により死亡する.成人型では10歳以上で骨格筋障害を来すが,心筋障害はまれである.
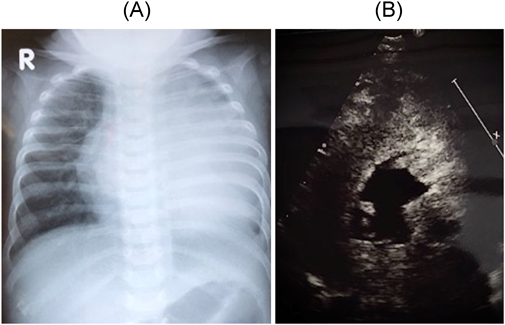
(A) Chest radiograph showing massive cardiomegaly. (B) Short-axis view of the left ventricle demonstrating severe concentric left ventricular hypertrophy on echocardiogram. Modified from ref 7).
新生児マススクリーニングにおいて乾燥ろ紙血でGAA酵素活性が測定されている.欠損疑い例はリンパ球または培養皮膚線維芽細胞によるGAA酵素活性や遺伝子検査,骨格筋生検により確定診断する.筋病理では空胞病変,グリコーゲン蓄積,酸フォスファターゼ活性の上昇がみられ,生検筋のGAA酵素活性の低下で証明することもできる.
ポンペ病遺伝子検査(かずさ遺伝子検査室),対象遺伝子:GAA,保険点数3,880点
乳児期の早期GAA酵素補充療法は生後5か月未満の乳児において心筋症の進行抑制がみられており,早期診断が極めて重要である8).
ライソゾーム病(自己貪食空胞性ミオパチー)
X連鎖遺伝.男女ともに発症し,男性は,心筋症,ミオパチー,知的障害が三大症状である.女性は主に心筋症のみで発症する.
ライソゾーム関連膜蛋白2型(LAMP-2)をコードするLAMP2遺伝子の病的バリアントにより引き起こされる.LAMP-2はライソゾーム膜の主要な糖タンパク質であり,LAMP-1とともにライソゾーム膜を埋め尽くしており,ライソゾーム膜をライソゾーム腔内の消化酵素から守っている役割を果たしているが,Danon病では骨格筋の免疫染色でLAMP-2蛋白が消失し,オートファジー機能低下により通常は骨格筋病理ではみられない自己貪食空胞が多数出現する.
ライソゾーム内の蓄積物質により肥大型心筋症様心肥大9)(Fig. 4)を認める.成人以降で拡張相肥大型心筋症へ移行し,心臓移植を行わない場合は25歳までに死亡し,生命予後不良である.心筋線維化が進行すると致死性不整脈から突然死に至るケースもある.心臓発生の過程においてもグリコーゲンの蓄積した心筋細胞が心房心室間の線維輪形成を阻害することが知られており,副伝導路が残存する.
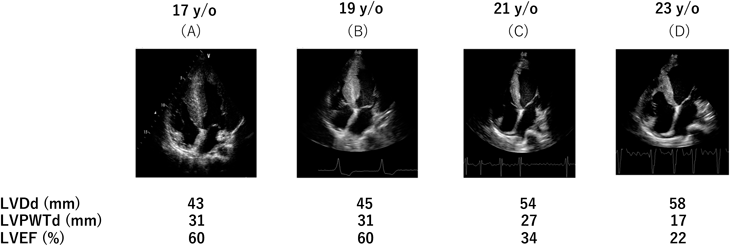
(A) Hypertrophic cardiomyopathy with normal LVEF of 60%. (B) Normal LVEF of 60%. (C) Dilated left ventricle with reduced LVEF of 34%. (D) Severely dilated left ventricle with reduced LVEF of 22%. LVDd, left ventricular diameter at end-diastole; LVEF, left ventricular ejection fraction; LVPWTd, left ventricular posterior wall thickness at end-diastole. Modified from ref 9).
男性では心筋症以外にミオパチー,精神遅滞を呈する.ミオパチーは近位筋優位の筋力低下と筋萎縮を認める.血清CK値は1000 IU/L程度に上昇しているため,高CK血症における鑑別疾患となりうる.精神遅滞は軽症の事が多い.女性は心筋症のみを発症する.
骨格筋生検で筋鞘膜の性質を有する自己貪食空胞の存在が特徴的であり,この空胞膜はジストロフィン関連タンパクが発現し,アセチルコリン・エステラーゼ活性を有する.したがって,筋病理上で自己貪食空胞を伴う筋線維,および空胞膜上でのアセチルコリンエステラーゼ活性を証明することや免疫組織化学染色にてジストロフィン関連タンパクの発現を認めることが診断に重要である.電子顕微鏡像ではオートファゴソームを認め,自己貪食空胞周囲に基底膜の存在を認める.遺伝子検査にてLAMP-2遺伝子の病的バリアントを認める10).
ダノン病遺伝子検査(かずさ遺伝子検査室),対象遺伝子:LAMP2,保険点数3,880点
対症療法主体であり,心不全や致死性不整脈に対してβ遮断薬,植込み型除細動器の適応を考慮する.心不全進行例に対しては心臓移植適応となりうるが,心外合併症の程度により慎重に判定する必要がある.
有機酸代謝異常症
常染色体潜性遺伝(劣性遺伝)
プロピオニルCoAカルボキシラーゼ(PCC)をコードする遺伝子PCCA, PCCBの病的バリアントにより発症する.PCCはミトコンドリア内でプロピオニルCoAをメチルマロニルCoAへ変換する酵素であるが,メチルマロニルCoAは最終的にスクシニルCoAへ変換されTCA回路に入る.プロピオニルCoAは奇数鎖脂肪酸やバリン,イソロイシンなどの分枝鎖アミノ酸の中間代謝物質で蓄積するとミトコンドリア内の様々な酵素を阻害し細胞毒性を有する.生体はプロピオニルCoAをカルニチンと結合し細胞内より除去し尿中へ排泄する.そのため二次性のカルニチン欠乏を起こして心筋をはじめ肝,脳,骨格筋などの臓器障害を呈する11).メチルマロニル-CoAからコキシニル-CoAへの変換を触媒するメチルマロニル-CoAミューターゼ(MMUT)の機能障害によってもプロピオン酸塩障害を呈する(メチルマロン酸血症).
遅発型では二次性のカルニチン欠乏により拡張型心筋症を来す.またQT延長症候群を認めることが知られている.
重症型は新生児期に急性代謝不全症状で発症し,重度の代謝性アシドーシス,高アンモニア血症を引き起こす.遅発型では乳幼児期から繰り返す嘔吐,精神運動発達遅延を認める.
血中アシルカルニチン分析でプロピオニルカルニチン(C3)の上昇が認められる.アセチルカルニチン(C2)との比(C3/C2比)の上昇は本症に特異的である.遺伝子検査はPCCのαサブユニットをコードするPCCA遺伝子およびβサブユニットをコードするPCCB遺伝子のいずれかの病的バリアントを同定できれば確定診断となる.
プロピオン酸血症遺伝子検査(かずさ遺伝子検査室),対象遺伝子:MMUT,PCCA,PCCB,保険点数8,000点
カルニチン欠乏に対してL-カルニチンを投与する.乳児では分枝鎖アミノ酸除去ミルクを併用し,タンパク摂取制限を開始する.
オルガネラ病
原因遺伝子が核DNAおよびミトコンドリアDNA上に300個以上あり,全ての遺伝形式を有する.
心筋ミトコンドリアの構造や機能に関わる遺伝子異常を背景とし,ミトコンドリア内膜に存在する呼吸鎖酵素複合体が十分に機能せずATP産生障害が起こることにより心筋症を発症する12).
肥大型心筋症様心肥大,拡張型心筋症,左室心筋緻密化障害など様々な表現型を呈し,新生児・乳児期早期発症例では哺乳不良や代謝性アシドーシスを伴い,いずれも生命予後は不良である.幼児,学童期以降では神経筋疾患,代謝性疾患に合併する二次性心筋症の鑑別において診断されることがあり,成人以降に心不全を発症することがある.全身のミトコンドリア病に合併する心筋症としてはMELASが代表的で,ミトコンドリアDNAの点バリアント(m.3243A>G)を認め,40%程度に肥大型心筋症様心肥大を合併し,進行例では心伝導障害と拡張相肥大型心筋症を認める.新生児・乳児における男児の重症心不全例ではBarth症候群の可能性を常に念頭においておく必要がある.Barth症候群はミトコンドリア内膜のカルジオリピン成熟障害によって発症するミトコンドリア病で,X連鎖遺伝形式で男児に発症し,左室心筋緻密化障害,好中球減少,ミオパチー,成長障害,3-メチルグルタコンサン尿症を特徴とする.
中枢神経症状,ミオパチー,眼症状,肝機能障害,腎障害,糖尿病,血液疾患,難聴,低身長など多彩な表現型を示す.心外合併症に乏しく心筋症孤発例の場合は診断に苦慮することがある.
ミトコンドリア心筋症の確定診断はBernierの診断基準に従い,(1)組織生化学検査,(2)病理学検査,(3)遺伝子検査を実施する.組織生化学検査は,生検,剖検で採取された心筋組織の呼吸鎖酵素活性を測定することで確定診断が可能である.病理学検査では電子顕微鏡像でミトコンドリアの著明な増加を認める他,ミトコンドリアの大型化,クリステ形態の異常が認められる.光学顕微鏡用試料作製時のパラフィン包埋ブロックがあれば呼吸鎖酵素複合体に対する抗体を用いた免疫染色法が有用である.本法はComplex I,II,IVのサブユニットに対する一次抗体を用い,本症に多いComplex IまたはIVの染色性の低下を証明する(Complex IIは内因性コントロールで使用)ことで診断し,Complex I/Complex II比,あるいはComplex I/Complex IV比が正常コントロールの30%以下であれば呼吸鎖酵素複合体欠損と判断し,組織生化学検査の代替診断として使用している(Fig. 5)13).ミトコンドリア病遺伝子パネルシーケンスによる遺伝子検査が令和4年度診療報酬改定(厚生労働省)において保険収載され,契約施設からの検査依頼受付が開始されている.ミトコンドリア病遺伝子パネルには,核のDNAにコードされた367個の原因遺伝子のほか,ミトコンドリアDNAの全周(16.6 kb)も対象としており,新生児や乳児のミトコンドリア病疑い例に対して罹患組織の生検が困難な例や病理,組織生化学的にミトコンドリア病と診断された例においては遺伝子検査を行って確定診断する.
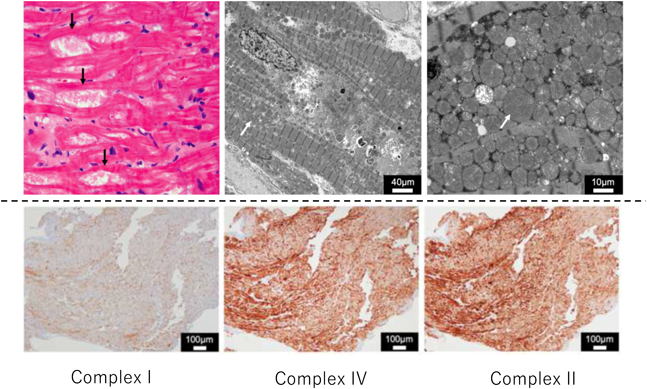
(Upper panel) Light microscopy image of the biopsied right ventricular myocardium showing cytoplasmic vacuolization (black arrow) (left panel). Electron microscopy of the biopsied right ventricular myocardium shows marked proliferation of mitochondria (white arrow) within the myofibrils (middle and right panels). (Lower panel) Immunohistopathological analysis of the biopsied myocardium using antibodies against respiratory chain enzymes. The left, middle, and right panels show the immunohistochemical analysis of complex I, IV, and II, respectively. The signal intensity of complex I compared with complex II was 22% of that of normal, and that of complex IV compared with complex II was 116% of that of normal. Modified from ref 15).
ミトコンドリア病遺伝子検査(順天堂大学),対象遺伝子:367原因遺伝子(現在は契約施設からのみ依頼を受け付けている)
根本治療は現在確立したものはない.全身の急性ミトコンドリア障害の際に生じる急性心不全増悪に対してはビタミンカクテル療法が有効なことがある.慢性心不全に対してはレニン・アンギオテンシン系阻害薬やβ遮断薬を用いた一般的な心保護療法を行う.心室頻拍,心室細動などの致死性不整脈で発症することもあり,電気的除細動やアミオダロン,Ia群薬を考慮する.発症後の二次予防としてICD植込術は適応となるが,突然死予防を目的とした一次予防でのICD植込は行われていない.
先天異常症候群
常染色体顕性(優性)遺伝 ごく一部に常染色体潜性(劣性)遺伝
RAS/MAPK経路は細胞の増殖,分化,代謝などに関連する細部内シグナル伝達経路のうちの1つでPTPN11, RAF1, RIT1, SOS1など様々な遺伝子が関与している.このシグナル伝達経路に関わる病的遺伝子バリアントによって発症する疾患群をRASopathiesと呼称し,代表的な症候群であるNoonan症候群をはじめ,多発性黒子を伴うNoonan症候群,Costello症候群,Cardio-facio-cutaneous(CFC)症候群などがある.二次性心筋症として重要な心合併症は肥大型心筋症様心肥大である.RASopathiesの心肥大メカニズムは心筋細胞の肥大と考えられていたが,心筋病理像から心筋細胞過増殖を示す証拠もみつかってきている.
乳児期早期から肥大型心筋症様心肥大を認めることがあり,発症年齢は鑑別診断において重要である.主にPTPN11, RAF1, RIT1などの遺伝子に病的バリアントを認める.遺伝子型によって肥大型心筋症の表現型に特徴があり,PTPN11では非対称性の心筋肥大が多いのに対してRAF1では全周性肥大が多い.閉塞性肥大型心筋症の臨床像を呈することも多い.乳児期早期発症の予後は不良で2歳時の生存率は30%である.
肥大型心筋症以外の心合併症としては肺動脈弁狭窄,心房中隔欠損症などがある.
特徴的な顔貌(眼瞼下垂,眼間乖離,内眼角贅皮,眼裂斜下,鞍鼻,耳介低位),翼状頸,胸郭異常(漏斗胸,鳩胸),関節過伸展,側弯を認める.
50~70%の症例で低身長を認め,GHホルモン療法の保険適応がある.重要な心外合併症に先天的なリンパ管形成異常があり,難治性の乳び胸や末梢リンパ浮腫を合併する.
軽度の精神運動発達遅滞を認める.
臨床症状より本症を疑い,遺伝子検査(下記)で診断確定する.
ヌーナン症候群遺伝子検査(かずさ遺伝子検査室),対象遺伝子:PTPN11, SOS1, RAF1, RIT1, KRAS, NRAS, SHOC2, CBL, BRAF, SOS2, MRAS, RRAS, LZTR1, RRAS2,保険点数8,000点
コステロ症候群遺伝子検査(かずさ遺伝子検査室),対象遺伝子:HRAS,保険点数5,000点
CFC症候群遺伝子検査(かずさ遺伝子検査室),対象遺伝子:KRAS, BRAF, MAP2K1, MAP2K2,保険点数5,000点
左室流出路狭窄に対してβ遮断剤やシベンゾリンが投与される14).狭窄が強いか,薬剤に抵抗性の場合は外科的中隔心筋切除術,あるいは経皮的中隔心筋焼灼術の適応である.
神経筋疾患
X連鎖遺伝.男性のみに発症する疾患で女性は保因者となるとされてきたが,女性(ヘテロ接合体)でも症状が発現することが報告されている.
ジストロフィン蛋白をコードするDMD遺伝子の病的バリアントにより骨格筋,心筋における筋線維の変性,壊死を主な病変とし,進行性の筋力低下や呼吸不全,心機能不全を来す.Duchenne型は10歳までにほぼ歩容消失するが,Becker型は成人以降でも歩行は可能である.
Duchenne型(DMD)/Becker型(BMD)のいずれも拡張型心筋症類似の心筋症を発症し,致死的経過をたどることがある.他の拡張型心筋症とは異なり,初期の段階では心拡大を伴わない心収縮障害から始まる.早ければ10歳代から進行するが,左室駆出率異常の平均発症率は14.3歳であることが報告されている.DMDでは歩容消失のため心不全症状がわかりにくく,心エコーや心臓MRIなどの画像診断によって診断される.心筋の置換性線維化は通常左室側壁心外膜側から起こり,経年性に全周性および貫壁性に進行する.限局性心筋障害の段階では心不全症状はなく,心エコー図による左室駆出率は正常であるが,線維化が広範囲に及ぶと駆出率は低下しはじめ,その後左室拡大が進行する.
心臓MRI検査は体格や側弯などの姿勢異常に制限されずに全体的な左室機能と壁運動評価が可能である上,遅延造影像やT1マッピング法による心筋組織性状を評価できるためDMD心筋症管理において大変重要な役割を担っている.
DMDでは進行性の筋力低下により10歳までにほぼ歩容消失し,呼吸筋の筋力低下により呼吸機能障害が進行する.その他,脊柱筋の筋力低下による側弯症や咽頭筋障害による摂食・嚥下障害が出現する.
上記運動機能低下や血清CK値の上昇から本症を疑い,遺伝子検査(下記)によって確定診断する.
MLPA(Multiplex Ligation Probe Amplification)法でエクソン単位の欠失・重複を判定する(保険適応).この方法でDMD/BMD患者の7割において診断可能である.MLPA法で異常が検出されない場合は筋ジストロフィー遺伝子検査(かずさ遺伝子検査室)に進み,微小変異の同定を行う(保険適応).
筋ジストロフィー遺伝子検査(かずさ遺伝子検査室),対象遺伝子:DMD,保険点数3,880点
心筋症の発症や心機能低下抑制のためにACE阻害薬,β遮断薬が用いられる15).
一部のDMDにおける骨格筋へのアンチセンス核酸製剤によるエキソンスキッピング療法が始められているが,心筋症の発症抑制効果は報告されていない.
小児の原因不明な心筋症の鑑別目的に心筋生検が行われる(AHA/ACCF/ESC Scientific Statement, Class IIa).二次性心筋症の鑑別において光学顕微鏡所見では空胞病変を認めることがあるが,これらは細胞内の複合脂質がホルマリンで化学固定されずに可視化されなかったものである.電子顕微鏡所見では四酸化オスミウムで脂質を化学固定するためそれらの一部は可視化され,Fabry病では同心円状層構造物(Zebra body),Danon病ではオートファゴソーム,ミトコンドリア心筋症ではミトコンドリアの著明な増加として認識できる.また,免疫染色法を用いて筋ジストロフィー症やミトコンドリア心筋症の鑑別診断を行うことも可能である.採取した心筋は病理のみならず各種酵素活性や質量分析,mRNA発現,ウイルスゲノム検索,細胞培養など目的に応じてさまざまな検査に活用できる(研究目的に人の検体を扱う場合は予め施設内の倫理審査を通しておく必要がある).注意したいことは,検査の目的によって採取した心筋組織の処理が異なることである.光学顕微鏡用には10%ホルマリン,電子顕微鏡用には2.5%グルタールアルデヒド,ウイルスゲノム検索や酵素活性測定の場合は未処理のまま−80度で凍結保存しておかねばならない.
二次性心筋症は原因となる背景疾患の鑑別が最も重要である.詳細な家族歴の中で原因がみつかる場合や,心臓以外の症状や臓器障害が決め手となる場合も多く,眼科的スクリーニングや聴力検査,神経学的検査など合併症の有無に留意する.典型的な表現型によっては遺伝子検査によって確定診断できるが,心筋症孤発例の場合は心筋生検を行い,病理学的な鑑別を進める.光学顕微鏡では空胞病変を認めることが多いが,この空胞は電子顕微鏡では可視化できるので診断につながることがある.生検した心筋組織は病理検査のみならず各種酵素活性やウイルスゲノム検索などに用いることもできるので,採取後の保存方法をあらかじめ決めておくことが望ましい.二次性心筋症の中にはすでに治療が可能となっている疾患があり,早期診断法の開発と発症予防が今後は大いに期待される.
本稿について,開示すべき利益相反(COI)はない.
1) Maron BJ, Towbin JA, Thiene G, et al: American Heart Association; Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; Council on Epidemiology and Prevention: Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006; 113: 1807–1816
2) Elliott PM, Anastasakis A, Borger MA, et al: Authors/Task Force members: 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35: 2733–2779
3) 日本心不全学会合同ガイドライン:心筋症診療ガイドライン(2018年改訂版).2019
4) Kubo TKH, Kitaoka H: Imaging of left ventricular hypertrophy: A practical utility for differential diagnosis and assessment of disease severity. Curr Cardiol Rep 2017; 19: 65
5) Linhart A, Palecek T: Narrative review on Morbus Fabry: Diagnosis and management of cardiac manifestations. Cardiovasc Diagn Ther 2021; 11: 650–660
6) 日本先天代謝異常学会:ファブリー病 診療ガイドライン2020.2020
7) Senarathne UD, Jasinge E, Viknarajah Mohan S, et al: Non-specificity of symptoms in infantile-onset Pompe disease may delay the diagnosis and institution of treatment. BMJ Case Rep 2022; 15: e247312
8) 日本先天代謝異常学会:ポンペ病診療ガイドライン2018.2018
9) Wang S, Wang Q, Zhai N, et al: Progression of Danon disease with medical imaging: Two case reports. J Int Med Res 2021; 49: 300060520986676
10) 日本神経学会:自己貪食空胞性ミオパチー診療の手引き.2019
11) Deodato F, Boenzi S, Santorelli FM, et al: Methylmalonic and propionic aciduria. Am J Med Genet C Semin Med Genet 2006; 142C: 104–112
12) 武田充人:ミトコンドリア心筋症.日小児循環器会誌2017; 33: 287–296
13) Takeda A, Murayama K, Okazaki Y, et al: Advanced pathological study for definite diagnosis of mitochondrial cardiomyopathy. J Clin Pathol 2021; 74: 365–371
14) Gelb BD, Roberts AE, Tartaglia M: Cardiomyopathies in Noonan syndrome and the other RASopathies. Prog Pediatr Cardiol 2015; 39: 13–19
15) Feingold B, Mahle WT, Auerbach S, et al: American Heart Association Pediatric Heart Failure Committee of the Council on Cardiovascular Disease in the Young; Council on Clinical Cardiology; Council on Cardiovascular Radiology and Intervention; Council on Functional Genomics and Translational Biology; and Stroke Council: Management of Cardiac Involvement Associated With Neuromuscular Diseases: A Scientific Statement From the American Heart Association. Circulation 2017; 136: e200–e231


This page was created on 2024-12-02T18:04:16.277+09:00
This page was last modified on 2025-03-10T16:33:20.000+09:00
このサイトは(株)国際文献社によって運用されています。