診断の契機
失神や心肺蘇生例の原因精査を契機に診断される場合がある.本邦における小児期の院外心停止例における原因疾患として,LQTSは先天性心疾患に次いで多かった6).また特に間代性でない痙攣発作を生じた症例で原因疾患の鑑別に挙げる必要がある7).本邦には学校心臓検診があるため,無症状で診断される症例が多いことも特徴である3, 8).なお胎児不整脈を契機に診断される症例もあるが,こちらは他稿に譲る.
診断基準
2013年のHRS/EHRA/APHRS Expert consensus statementの診断基準を参照する(Table 2)9).なおLQTSリスクスコアは,Schwartzらの考案した臨床診断スコア(Table 3)10)であるが,このリスクスコアの臨床的特徴に留意した病歴聴取も重要である.
Table 2 Expert consensus recommendation on LQTS diagnosis9)| 1. LQTS is diagnosed: |
| a. In the presence of an LQTS risk score ≥3.5 in the absence of a secondary cause for QT prolongation and/or |
| b. In the presence of an unequivocally pathogenic mutation in one of the LQTS genes or |
| c. In the presence of a QT interval corrected for heart rate using Bazett’s formula (QTc) ≥500 ms in repeated 12-lead ECGs and in the absence of a secondary cause for QT prolongation. |
| 2. LQTS can be diagnosed: |
| a. In the presence of a QTc between 480–499 ms in repeated 12-lead ECGs in a patient with unexplained syncope in the absence of a secondary cause for QT prolongation and in the absence of a pathogenic mutation. |
Table 3 LQTS risk score (LQTS diagnostic criteria)10) | Points |
|---|
| Electrocardiographic findingsa | |
| A | QTcb | ≥480 ms | 3 |
| 460–479 ms | 2 |
| 450–459 (male) ms | 1 |
| B | QTcb | 4th minute of recovery from exercise stress test ≥480 ms | 1 |
| C | Torsade de pointesc | 2 |
| D | T-wave alternans | 1 |
| E | Notched T-wave in three leads | 1 |
| F | Low heart rate for aged | 0.5 |
| Clinical history |
| A | Syncope | With stress | 2 |
| Without stress | 1 |
| B | Congenital deafness | 0.5 |
| Family history |
| A | Family members with definite LQTSe | 1 |
| B | Unexplained sudden cardiac death below age 30 among immediate family memberse | 0.5 |
| SCORE: |
| ≤1 point: low probability of LQTS |
| 1.5 to 3 point: intermediate probability of LQTS |
| ≥3.5 points: high probability |
aln the absence of medications or disorders known to affect these electrocardiographic features.
bQTc calculated by Bazett’s formula where QTc=QT/√RR.
cMutually exclusive.
dResting heart rate below the 2nd percentile for age.
eThe same family member cannot be counted in A and B. |
各遺伝型の病態と不整脈の契機
各遺伝型の病態を理解していると,心イベントの契機と生活管理,検査結果の解釈,治療選択をより適切に行える.特に頻度の高いLQT1,2,3の病態は以下の通りである.
LQT1は緩徐活性型遅延整流性カリウム電流(IKs)の低下が原因である.このイオンチャネルは交感神経刺激により電流量が大きくなるという性質と,脱活性化が遅いため頻拍時に蓄積効果により電流量が大きくなるという性質がある11).LQT1ではIKsチャネルの機能低下により,運動等によって交感神経が緊張し頻拍となった際に,適切なIKs増加が得られないため,QT延長に関連した不整脈が惹起される.
LQT2は急速活性型遅延整流性カリウム電流(IKr)の低下が原因である.LQT2モデルにおいて,イソプロテレノール投与により活動電位は一過性に延長した後に短縮した.イソプロテレノール投与開始直後には,L型カルシウム電流(ICa,L)増加がIKs増加より早く生じるため,IKrの再分極への寄与がより大きくなるが,LQT2ではIKrが減少しているためにこの一過性の延長を生じたと考えられた12).この機序によりLQT2では安静時に急な交感神経緊張がある場合,例えば安静時にアラームで驚く等によりTdP発生の危険性が高くなると考えられる.また低カリウム血症ではさらにIKrが減少するため,TdPが惹起され易くなる13).
LQT3はINaチャネルの不活性化障害によりlate Na電流(INa-L)が増加することで機能亢進を来すことが原因である14).さらに,徐脈時にはINa-Lが増加し15)QT延長が増悪するため,LQT3では睡眠中などの徐脈時に心室性不整脈を起こしやすい16).LQT3と同等ではないが,この機序による徐脈依存性のQT延長は他の遺伝型でも共通している16).
心外合併症
LQTSでは心外合併症を生じる場合がある.KCNQ1またはKCNE1のホモ接合性変異や複合ヘテロ接合性変異では聾を伴うJervell and Lange-Nielsen症候群(JLNS)を来し得る17).LQT7(Andersen–Tawil症候群)はKCNJ2遺伝子変異が原因となり,周期性四肢麻痺,特異的顔貌や第5指の彎曲等の身体的特徴を有する18).著明なU波によるQU間隔の延長を認め,LQTSとは異なる疾患とする考えもある.LQT7では心外合併症が揃っていない場合も多い19).LQT8はCACNA1C遺伝子変異を原因とするが,さらに合指症,精神神経症状,特異的顔貌等の心外合併症がある場合をTimothy症候群(TS)と診断している20, 21).精神神経症状としては特に自閉症が多いが,重度の精神運動発達遅滞やてんかんも合併し多様である.また合指症を認める症例は一部に限られる.
安静時12誘導心電図
①QT間隔の計測方法
接線法によるマニュアル計測が推奨される.接線法ではIIあるいはV5誘導で計測し,T波下降脚の最大傾斜部分に接線を引き,P波直前(困難な場合はQRS波直前)を結んだ基線と交差する点をT波の終点とする22).自動計測で多く用いられている微分法では,接線法よりQT間隔が長く計測されやすいが,逆にT波終末部の認識の誤りにより過小評価される場合もある.特にT波形態に特徴のあるLQTSにおいては自動計測とマニュアル計測の値に差があることが多い23).このため自動計測により抽出された場合は接線法によるマニュアル計測が必要であり,また自動計測で正常と判断された場合でも医師による判読が必要である.
②補正QT間隔とその評価
計測したQT間隔は,心拍数(先行RR間隔)で補正して評価する.補正式は数種類あり,算出される補正QT間隔(QTc)は式により異なる23, 24).このため,評価の際は参照する既存データと同じ補正式を使用する.例えば,LQTSリスクスコア10)ではBazettの補正式(QT間隔/RR間隔1/2),学校心臓検診の1次検診での判定25)ではFridericiaの補正式(QT間隔/RR間隔1/3)を使用する.本文中では,特に断りがなければBazettの補正式によるQTcとする.LQTSリスクスコアでは,全年齢で性別を問わずQTc>460 ms,特に思春期以降の男性ではQTc>450 msを有意な延長としている.健常者,LQTS患者ともに,QTcは年齢により変化し,さらに性別,遺伝型による変化の違いも示されているため26),今後小児期独自のLQTS診断基準が望まれる.学校心臓検診ではFridericia補正でのQTcで,小学1年男女430 ms以上,中学1年男女440 ms以上,高校1年男子440 ms/女子450 ms以上を1次検診での抽出基準としている25).
③T波形態の異常
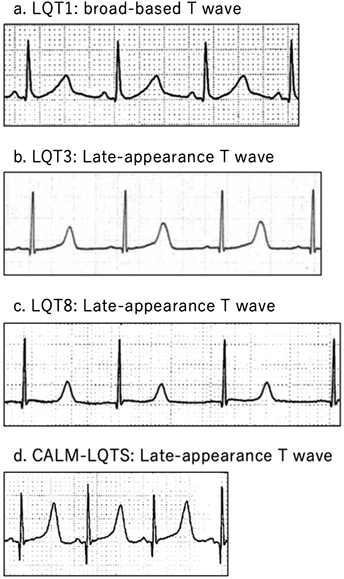
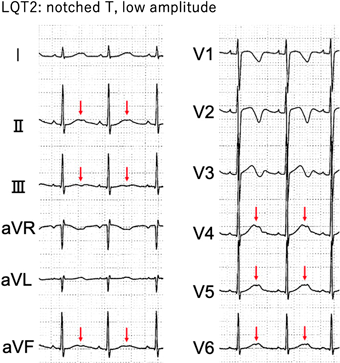
T波形態も診断と遺伝型の推定に重要である(Fig. 1, 2).LQT1では,broad-based T波が特徴的である.LQT2では,low amplitude T波に加えてnotched(あるいはbifid)T波を認めることがある.notched T波はLQTSリスクスコアでも評価する所見であるが,小児期には正常でも経時的なT波変化の過程でV2–4を中心にnotched T波らしい波形を示す場合がある.LQT2の典型例では左側胸部誘導や下壁誘導でもnotched T波を認める(Fig. 2).LQT3ではlate-appearance T波を認めるが,これは活動電位第2相(プラトー相)の延長に起因する所見である.LQT8,さらにカルモジュリン異常が原因となるCALM-LQTS(LQT14–16)では,L型カルシウム電流(ICa,L)の増加が主病態だが,LQT3と同様にプラトー相が延長し,late-appearance T波を認める27–29).
④洞性徐脈の評価
同じ遺伝型でも病的バリアントによる機能変化の違い等により,徐脈の有無は様々である30).またLQTSリスクスコアにおいて,徐脈は年齢ごとのデータでの2パーセンタイル未満とされており10),各年齢における心拍数のデータ31)を参照する.
二次性QT延長
成人と比較すると頻度は低いが,各種薬剤,徐脈,電解質異常(低K,低Mg,低Ca),甲状腺機能低下等による二次性QT延長の鑑別が必要である32).ただし,二次性QT延長と診断される症例の中に先天性LQTSの遺伝子に病的バリアントが同定されることもあり,1)40歳未満,2)二次的要因解除後のQTc>440 ms,3)心イベントの既往がある場合で病的バリアントの同定率が高く,KCNH2遺伝子の病的バリアントが多かったと報告されている33).また心肺蘇生後の患者では,おそらくカテコラミン心筋障害により,一過性にQT間隔が延長する場合があり注意する.T波陰性化を伴うことも参考になるが,最終的には経時的変化を踏まえて判断する34).小児期のアスリートでトレーニングに関連したQT延長を認めることが報告されている35).
負荷試験
①運動負荷試験
LQT1,2の小児例において安静時QTc<460 msの症例が46%あり,エルゴメーターを用いた運動負荷試験で,回復期7分のQTcが460 ms以上では感度86%・特異度96%でLQTSが診断可能であった36).さらにトレッドミルあるいはエルゴメーターを用いた成人例での検討で,安静時QTcはLQTS症例の25%で正常で,回復期4分のQTc>480 msでは感度36%・特異度100%,QTc>445 msでは感度90%・特異度90%で診断可能であった37).これを踏まえてLQTSリスクスコアに運動負荷後4分の項目が追加され38),回復期4分のQTc>480 msで1ポイントを加算している.なお,β遮断薬内服下でも結果の解釈への影響はなかったとされており36, 39),内服下での評価でも差し支えない.
運動負荷試験において,QTcの経時的な変化様式は遺伝型により異なることが報告されており,これを把握しておくことは非常に重要である.LQT1では運動中からQTcは延長し,回復期1分でも引き続き延長しているが,LQT2はそのタイミングではQTcは短縮している36).LQT1では運動後3分のQTcが安静時から30 ms以上延長している場合に75%の確立で予測可能であり39),安静時からの変化の大きさを確認することも有用である.LQT2では,回復期1分の短縮から徐々に延長し,安静時と同程度となる36).LQT1,2での変化の違いは,それぞれで低下しているIKs,IKrについて,活動電位への寄与の程度が運動による交感神経緊張や心拍数上昇に伴い変化するためと考えられる.最終的な解釈の際に,Azizらの報告36)にも示されているように,正常例でも回復期5分では安静時より20 ms程度QTcが延長すること,LQT1において回復期1分のQTc延長は回復期3分ほど大きくないことにも留意するとよりよい.なおLQT1の心イベントリスクが,運動負荷によるQT間隔の変化の大きさで予想できるというエビデンスは十分ではない.基本状態のIKsおよびカテコラミンに対する電流変化は変異体により異なっているが40–42),その差が必ずしも心イベントリスクと相関していないためと推察する.LQT3の変化様式はLQT2に類似しており,QTcは運動のピークでは安静時よりも短縮し,その後徐々に安静時と同程度の長さへ戻る39).LQT8もLQT2に類似の変化であったが43),対象が限定的であり今後追加検討が必要と考える.
②エピネフリン負荷試験
検査の意義は運動負荷試験と同様である44, 45).ただしエピネフリン負荷試験では偽陽性を示す症例があり34, 46, 47),ESC心室性不整脈患者の管理と心原性突然死予防ガイドラインではLQTS診断のための検査として推奨されていない48).ただし,年少者などの運動負荷試験が施行困難な症例では,遺伝型と表現型の整合性の確認のための検査として実施されることもある.
③起立負荷試験
臥位から急に立位となる際の心拍数とQT時間,T波形態の変化を評価する49).現在は成人,小児とも診断のための検査として推奨はされていない50, 51).急な起立時に心拍数上昇がQT短縮より鋭敏に生じるため,正常例においてもQTcは成人で50 ms,小児(平均10歳)で80 ms延長するが52, 53),一般的に心電図評価の際にはこの急な心拍数上昇時のQTc延長があることに注意が必要である.
24時間Holter心電図
生活上の様々な変化を記録可能で,LQTSにおいてはT波交代減少(T wave alternans; TWA),TdPが非常に稀ではあるが同定されることがある(Fig. 3).TWAはT波形態が一拍ごとに交互に変化する所見で,再分極相の貫壁性電気勾配の変動を示している.この貫壁性電気勾配を基質にtriggered activityを契機にphase 2 reentryを生じTdPが発生し得る.したがって,TWAはTdP発生を予見する重要な所見である.LQTSリスクスコアには視覚可能なTWAが含まれているが,視覚不能なmicrovolt TWAもTdP発生と関連していると報告されている54).またHolter心電図での最大QTcが心イベント発生の予測に有用であったとする報告もある55).
臨床情報とこれまで述べた各種検査によりLQTSリスクスコアを評価できる.また心イベントの契機,安静時のT波形態と運動負荷試験回復期でのQTc変化様式を合わせた表現型から遺伝型が推測できている場合が多い.
遺伝学的検査
遺伝学的検査の適応については,2024年改訂版の心臓血管疾患における遺伝学的検査と遺伝カウンセリングに関するガイドライン56)を参照する.LQTSでは前述の通り必ずしも典型的な表現型を示すとは限らず,循環器医が強く疑う症例については遺伝学的検査の実施が望ましい.LQTSの遺伝学的検査は2008年に保険診療(遺伝学的検査8,000点,遺伝カウンセリング1,000点)として承認されている.
①原因遺伝子のバリアント同定
近年,次世代シークエンサー(Next generation sequencer; NGS)を用いた解析が主流であり,Table 1に示すDefinitiveの遺伝子を中心に,施設ごとに解析対象遺伝子が決められている.一般的に原因遺伝子に病的バリアントが同定されたLQTS患者の90%に,KCNQ1,KCNH2,あるいはSCN5Aの病的バリアントが同定される.商業ベースでは,頻度の高いこの3遺伝子のみ解析されている.ただし滋賀医科大学でのLQTS 17遺伝子の網羅的解析により,遺伝子診断症例の5.4%にLQT8/TSの原因遺伝子であるCACNA1C遺伝子の病的バリアントが同定されており57),late-appearance T波等の特徴から疑われる場合は,解析対象に含めることが望ましい.また乳児期から重症な表現型を示す症例ではCALM1-3も解析対象に含めるべきである58).
②同定されたバリアントの病原性の判断
LQTS原因遺伝子にバリアントが同定された場合,まず病原性の有無を判断する必要がある.筆者はまずヒトゲノムについての最大のデータベースであるgnomAD(https://gnomad.broadinstitute.org)や日本人のデータベースであるTOGOVAR(https://grch38.togovar.org/)を参照し,健常人での同定頻度を調べている.次に既報のバリアントであれば,病原性についてバリアントと疾患についてのデータベースであるClinVar(https://www.ncbi.nlm.nih.gov/clinvar/)を参照する.もし新規のバリアントで病原性の評価がなされていない場合,ACMGガイドライン59)に従い病原性を判断する.評価項目の一つであるアレル頻度について,LQTSは1/2000人の疾患で,ヘテロ接合型変異で発症している場合(常染色体顕性遺伝)が多いため,アレル頻度0.00025以下であれば疾患に関与している可能性がある.また機能予測プログラムとして,複数の予測プログラムを統合し数値化するCADDスコア(https://cadd.gs.washington.edu)やREVELスコア(https://sites.google.com/site/revelgenomics/)が最も広く用いられている.一般的にCADDスコア>20,REVELスコア>0.5~0.75で病原性があると予測している.最近ではFlanklin(https://franklin.genoox.com/clinical-db/home)やVARSOME(https://varsome.com/)などのサイトも病原性評価に有用である.ところでアミノ酸の変化のない同義置換バリアントは原則良性であるが,日本人に多いKCNQ1 c.1032G>A p.A344Asplはスプラシング異常を来す変異であり注意が必要である60, 61).また病原性の判断と疾患重症度の予測は別に考えるべきで,この点は後述する.
遺伝型と表現型の整合性の確認が重要である.整合性が不十分な場合は未検出の遺伝子異常等がある可能性も示唆される.
病原性のある遺伝子変異の重症度評価
最初にナンセンス変異であるか確認する.ナンセンス変異は,各アミノ酸に対応していたコドンが,塩基置換やフレームシフトにより,翻訳を停止する終止コドンへ変化する変異である.ナンセンス変異のあるアレルから転写されたmRNAは,ナンセンス変異依存mRNA分解機構(nonsense mediated mRNA decay; NMD)により分解されるため翻訳されない62).つまり短いタンパク質が産生されるわけではなく,正常アレルのみからの翻訳となるためチャネルの発現量が原則50%へ減少し機能低下を来す.このように遺伝子産物の量的な異常により,機能を十分に果たせなくなる状態をハプロ不全と呼ぶ.
次にナンセンス変異ではない場合について考える.LQT1, 2の原因となるIKs,IKrチャネルはそれぞれ4量体であるため,ミスセンス変異の多くでは優勢阻害効果(Dominant-negative effect; DN)がある.例えば4量体を構成する4つのタンパクのうち無機能の変異タンパクが1つ含まれた場合の機能は3/4,2つの場合は1/2,3つの場合は1/4に低下しそうだが,実際には変異タンパクが正常タンパクを妨げることで,機能はそれぞれ3/4,1/2,1/4よりも低下する.このDNによって,ヘテロ接合型変異では総合的に50%より大きな機能低下を生じていることがある.このようにDNでは機能低下がハプロ不全より大きくなり得るため,臨床的に重症度がより高い63, 64).ただしミスセンス変異の機能変化の程度は,その遺伝情報を元にイオンチャネルとして機能するまでの過程(転写,翻訳,trafficking,イオンチャネル機能)への影響等により様々であり65),機能が50%以上残存している変異もある63, 64).ミスセンス変異における機能低下の程度は既報を確認する必要がある.
DNでのイオンチャネルの領域ごとの重症度比較について述べる.LQT1ではKCNQ1のC末端領域よりそれ以外の膜貫通領域や細胞質ループ領域(cytoplasmic-loop; C-loop)の病的バリアントの方が重症度が高いとされている63).さらにC-loopに重症度の高い病的バリアントが多いことが示されているが42),C-loop以外の領域(C末端領域を含む)にも,重症度の高い病的バリアントはあることも念頭に置く66, 67).KCNH2では,DNでの領域ごとの重症度比較はこれまで報告がないが,pore領域にDNを来す病的バリアントが多く,DNとハプロ不全を区別せずに行われた検討ではpore領域に病的バリアントを有する場合が最も予後不良であった68).将来的には,病的バリアントごとのリスク層別化が期待されている.
複合ヘテロ接合性変異は遺伝子診断された患者の4~11%に認めるが,単一の病的バリアントと比較し重症であり,前述した通りKCNQ1またはKCNE1では聾を伴うJLNSを来し得る17).
最後に,病状を軽減したり増悪させたりする遺伝的要因や環境因子により,LQTSの表現型は多様となり得るため,同一家系内でも重症度が異なることがあることに留意する1).
QTc
QTc延長の程度は,心イベント発生率に有意に相関しており,最も重要なリスク因子である69–71).基本的には安静時心電図で評価を行う.QTc 500 ms以上で心停止や心臓突然死の危険性が特に上昇したと報告されている70).また心イベントの既往がある場合,QTcによらず再発のリスクが高い72).
経時的変化
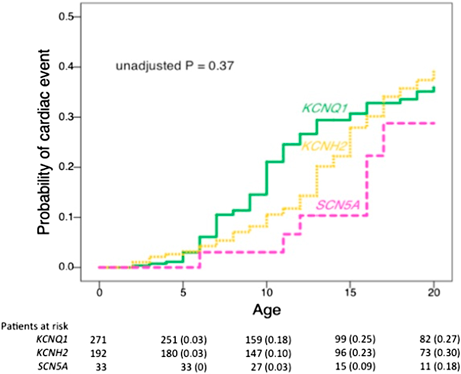
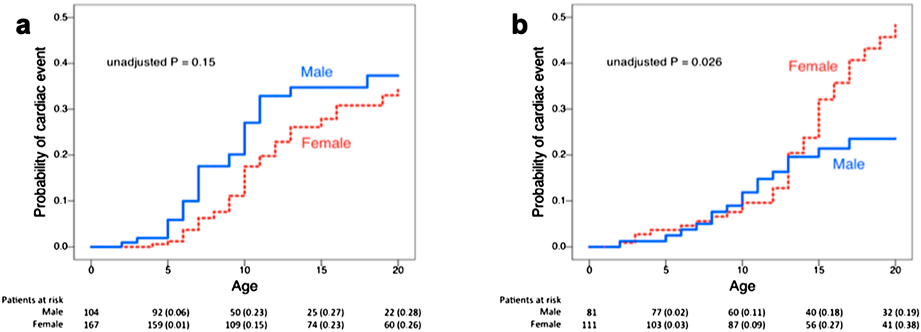
小児期の診療では,遺伝型,性別により経時的に不整脈の危険性が変化することにも注意する.Fig. 4, 5は,滋賀医科大学のコホートデータ73)であるが,米国のコホート74)でも同様の傾向が示されている.
LQT1では,就学頃から初発の心イベント発生頻度が増加し,思春期以降は減少する.学童期の男児に特に多いのは,より活動的で心イベント発生の契機が多いためと推測される.また男性においてテストステロン値が高いとQTcが短縮することが知られており75),テストステロン増加が心イベント抑制に寄与している可能性がある.一方LQT2では,13歳頃から女児の心イベントが急激に増加している.日本人女児では,思春期発来(エストロゲンの上昇)は10.0±1.4歳76),初潮は12.4±1.0歳で生じるが77),13歳頃からの心イベント増加を考慮すると月経周期に心イベントリスクの高い時期がある可能性がある.LQT2女性においては,プロゲステロン高値,特にプロゲステロン/エストロゲン比が高いほうがQTcは短く,卵胞期の初期が最もQTcが延長していた78).ただし同時期に心イベントが多いのかは不明で,今後の検討が期待される.LQT3では,性別によらず15歳頃から心イベントが増加するが,要因ははっきりしていない.私見だが心拍数の低下傾向が一因かもしれない.また実験的にも,性ホルモンにはイオンチャネルの発現変化79–81),電気生理学的機能への直接作用があることが示されている82–85)が詳細は他稿をご参照いただきたい.
生活上の注意点,治療
基本的には学校心臓検診のガイドライン25),遺伝性不整脈の診療に関するガイドライン32),不整脈非薬物治療ガイドライン86)等を参照いただきたい.以下,特に注意するべき点について述べる.
A. β遮断薬の選択,副作用
予防治療としてβ遮断薬が最も多く使用されるが,特にナドロールの有効性が高いことが複数のレジストリー研究で示されており87–89),ナドロールを第一選択薬とすることが多い.またプロプラノロールには,心イベントの再発抑制効果が他のβ遮断薬より劣っていたという報告があるが88),これはプロプラノロールが重症例の多い乳児例で使用されていたからと考察されている89).プロプラノロールは唯一,内服・静注製剤の両者があるβ遮断薬であり,その有用性は否定されないと考える.またアテノロールにもナドロールとほぼ同等の有効性があることを示した研究がある88).一方,メトプロロールの有効性については,肯定的な報告88)と否定的な報告がある87).
一般的にβ遮断薬により安静時のQT間隔はほとんど短縮しないが87, 90),プロプラノロールにはlate Na抑制作用があり他のβ遮断薬よりQT間隔の短縮が大きいとされている91).β遮断薬は,LQT1では臨床的に心拍数上昇時のQT間隔および再分極のばらつきを示すTpeak−Tendを短縮することが報告されており92),実験的にも同様の機序でTdPを抑制することが示されている93).LQT2においても心拍数上昇時のQT延長,早期後脱分極,再分極のばらつきの増加を抑制し,TdP発生を抑制することが示されている93, 94).このように頻度の高いLQT1,2においてβ遮断薬の治療効果は主に心拍数上昇時に認められ,安静時のQT短縮は治療効果判定には重要ではない.
ところでLQT3では,徐脈により増悪する病態や,実験的にβ遮断薬に催不整脈作用がある可能性が示されたことにより93),β遮断薬の使用は避けられてきた.しかし近年,臨床的にはβ遮断薬に催不整作用はないことが報告され95),さらに心イベントの抑制に有効であることも示されている90, 95).ただし,電気生理学的機序の特殊性からLQT3とBrugada症候群の両方の表現型を示し得るSCN5A c.5350C>A p.E1784K保因者96)では,病状に応じた治療決定が必要である.
β遮断薬の副作用について,LQT1,2患児において,稀ながら低血糖が認められている.これは膵島にも発現しているIKsおよびIKrチャネルの機能低下とβ遮断薬による抑制のためインスリン分泌が増加することが原因と考えられており,特に乳幼児では注意が必要である97).
B. メキシレチンの有用性
メキシレチンはIb群のNaチャネル遮断薬で,INa-Lを抑制しLQT3患者においてQTcを短縮させ,致死的不整脈の発生を減少させる98).上述の通りINa-Lは他の遺伝型のQT延長にも寄与しているが,メキシレチンのQT短縮作用はLQT1,2でも示されており16, 99),これらの遺伝型でも不整脈抑制効果を有する可能性がある.さらにTS(LQT8)においても不整脈が抑制された症例が報告されており100, 101),今後更なる検討が期待される.