Helen B. Taussigは,その遺稿となった,脊椎動物の心臓発生に関する1988年の論文において,「進化に伴う“奇形”とは,臓器特異的な異常であり,これは遺伝子変異によるものであると,私は信じる.」と述べた1).PCR法が発表されたのはこの頃であり,先天性心疾患(CHD; Congenital Heart Disease)に関連する遺伝子変異が初めて報告されたのは,この数年後であった.CHDに対する遺伝学的解析は,新しい網羅的遺伝学的解析の手法である,次世代シーケンサー(NGS; Next Generation Sequencer)の登場によって,近年さらに加速している.遺伝子変異とCHDの関連について,個別の原因遺伝子探索が加速する一方,これにとどまらず,NGSを応用してその発症機構にも踏み込んだ研究が次々と発表されている.本稿ではCHDに対する網羅的遺伝学的解析の歴史と展望について概説する.
CHDは出生あたりおよそ0.4~1.2%において発症する,最多の先天性疾患である2, 3).CHDの成因は多因子遺伝とされ,遺伝子変異と環境因子が複雑に影響して,発症につながると考えられている2).疫学的には,CHD孤発例において,同胞の罹患率は2~5%と一般人口よりも高い4).CHD家族例において,一親等に罹患者がより多く,また,双生児や同胞での罹患率はさらに高いという報告がある5).遺伝的に近いほどCHDの再現率が高いという事実は,CHDが多因子遺伝によることを示唆するものである.一方で,単一遺伝子異常によってCHDを発症する症例も存在し,染色体異常に伴うCHDおよびメンデル遺伝形式をとるCHDは全体の20%(それぞれ11.9%, 7.4%)とされている6).
1990年代以降,PCR法とSanger法を組み合わせることで,短いDNA配列を効率的に解析できるようになると,様々な疾患で遺伝子変異が報告されるようになった.大家系において発症に関連する遺伝子型の,染色体上の位置を統計解析する連鎖解析法や,染色体の構造異常による先天異常症候群に対する解析などから,原因遺伝子の染色体上の位置を絞り込み,候補遺伝子を同定する手法が用いられるようになった.単一遺伝子変異でCHDを生じるものとして,1997年にHolt-Oram症候群の原因となるTBX5が,複数の家族に対する塩基配列解析によって初めて同定された7, 8).さらにCHD孤発例の原因遺伝子としては,1998年に房室伝導障害を伴う心房中隔欠損症(ASD; Atrial Septal Defect)家系に対する連鎖解析によって,NKX2-5が初めて同定されている9).その後もGATA4, MYH6, TBX20など,次々とCHD原因遺伝子が報告されてきた10).
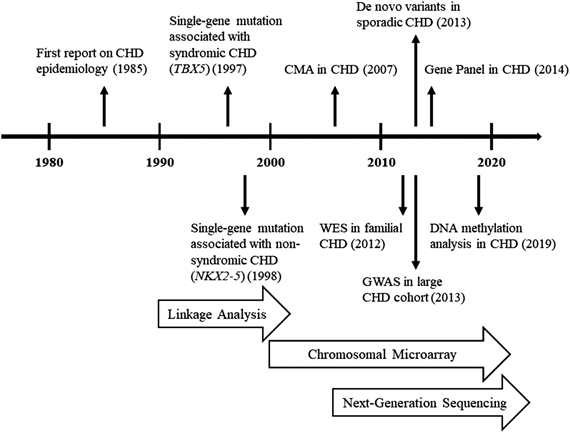
2003年に完了したヒトゲノム計画は,30億ドルの費用と13年の労力を掛けて,ヒトゲノムの全配列を発表した.これを嚆矢として,網羅的遺伝学的解析手法は急速な発展を遂げた.2005年に,ゲノム配列を網羅的かつ高速に解読できるNGS技術が実用化されてから,ヒトゲノムを解読するのにかかる費用は加速度的に低下している11).また,世界中で行われた網羅的遺伝学的解析の結果は漸次データベース化され,遺伝子研究の更なる加速に寄与している.NGS技術による塩基配列の解析を中心として,新たな技術が次々と登場することで,CHDに対する遺伝学的解析は発展を続けている(Fig. 1).
CHDに対する最初の網羅的遺伝学的解析の報告は,2012年にASDなどを含むCHD大家系に対して,NGSによる全エクソーム解析(WES; Whole Exome Sequencing)を行ったものである10).解析は既知の候補遺伝子から,ナレッジベースで原因遺伝子を探索するもので,候補としてMYH6遺伝子が挙がったものの,家系情報とは一致せず,原因遺伝子は不明で終わっている.世界中の研究機関が協力して,様々な人種を含む1,092人の全ゲノムデータ“1000 genome project”の成果が公開されて間もない時期である12).ゲノムデータベースが整備されたことで初めて,研究者が自ら膨大なコントロールを用意することなく,遺伝子研究を行うことが可能となった.その後もCHD家族例や症候群などにおいて,NGSによる既知の原因遺伝子を同定する試みが,数多く報告されている13–15).
全エクソームまたは全ゲノムを対象にしたNGS解析は高額な検査であるが,DNAの特定の部分だけを対象にした「遺伝子パネル」解析は,比較的低いコストで実施できる.2014年にはCHD関連の既知57遺伝子のエクソーム(蛋白コード部分)に対して遺伝子パネルを設計して,CHD 16家系に対して原因遺伝子を同定する試みが報告された16).原因遺伝子が同定されたのはCHD 9家系13変異(家族例の31%)であり,未知のCHD関連遺伝子が数多く存在することが示唆される一方で,パネル解析の診断能力について,その臨床応用に将来性が示された.注目すべき点として,心外病変が軽微であるにもかかわらず,原因遺伝子から既知の症候群と判明した数家系が認められ,臨床症状の多様性が明らかになった疾患があったことが挙げられる.その後も,候補遺伝子に対する解析は数多くの報告があるが,多数の遺伝子パネルを用いても,診断率は31~46%程度である16–18).遺伝子パネル解析は,全ゲノム解析や全エクソーム解析と比較すると安価であるという利点がある.サンプルあたりの解析費用は急速に低下しており,現時点で全ゲノム解析が約1,700ドル,全エクソーム解析が約800ドル,遺伝子パネル解析が約300ドルである19).また,遺伝子パネル解析は,特定の疾患における既知の原因遺伝子のみを解析対象とするため,解析が容易な手法である.今後WESなどの網羅的遺伝学的解析手法が普及するにつれて,候補遺伝子のパネルが更に充実していくことが期待される.
既知のCHD候補遺伝子に対するパネル解析の診断率は未だ高くないため,網羅的解析であるWESによって未知のCHD関連遺伝子が発見されることが期待されている.費用やデータ解析の難しさからは,全ゲノムを標的とした網羅的解析よりも,DNAの2%未満であるエクソームだけを標的とすることは,ほとんどのメンデル遺伝性疾患では蛋白コード部分に異常があることを考えれば妥当と考えられる11).2018年,CHD 30家系におけるWESを用いた研究では,3家系(10%)において,既知のCHD関連遺伝子に病的変異が同定されている20).
WES解析によって,新たなCHD症候群も報告されている.2017年にNature Genetics誌に掲載された論文では,指の拘縮などを伴う複数のCHD家系において,WESにより同定されたABL1遺伝子変異が,機能解析を含めて報告されている21).このABL1変異によるCHD症候群は,我々の研究室でも一家系を同定しており,この変異がABL1蛋白のキナーゼ活性を亢進させる機能獲得型変異であることを示すとともに,リン酸化プロテオーム解析を行い,ABL1蛋白のキナーゼ活性の亢進によって,どのような蛋白のリン酸化が影響を受けるかを解析した22).その結果,心疾患については22q11.2欠失症候群に関連するUFD1のリン酸化の亢進,指の拘縮や発育不全にはそれぞれAXIN1, ATRXのリン酸化の亢進が関与していることが示唆された.すなわち,ABL1変異により,様々な蛋白においてリン酸化が亢進することで,CHDを含めた多様な臨床症状の発症に至る機序が推定された.
CHD分野における,WESによる原因遺伝子変異の同定率は,高くない.様々な遺伝子をノックアウトしたマウスのうち,CHDを生じる割合から推計すると,500個を超える遺伝子が心臓発生に必須と推計されており,現時点では未知のヒトCHD関連遺伝子が数多く残されていると思われる23).また,CHD関連とされる候補遺伝子は,文献によって90~1,700個と幅がある24).これは,複数の症例で証明された変異のみという厳密なクライテリアから,動物や細胞実験のみを根拠とする変異まで,様々なレベルのエビデンスが混在しているためである.ヒトにおいて検出された遺伝子変異の病原性は,最終的には蛋白解析や網羅的RNA解析,動物実験などの方法で証明される必要がある25).
今後もWESなどの網羅的遺伝学的解析が,稀少なCHD家系や症候群に実施されることで,CHD関連遺伝子は増えていくことが予想される.こうした未知の遺伝子に対して,良質な機能解析などの追試が実施されることで,確固たるエビデンスが蓄積していくことが望まれる.
ある疾患に対する健常コントロールとなるべき,ヒトゲノムデータベースが整備されていなかった時代には,おおむね100人程度の健常コホートに当該遺伝子変異が認められないことをもって,ある遺伝子変異の病原性の根拠とされた26, 27).メンデル遺伝性疾患の場合,表現型が致命的であるほど,健常コホートに当該遺伝子変異がある可能性は低いためである.しかし,現代の数千人規模の大規模コホート研究の結果を用いて,既報の遺伝子変異を再検証すると,健常人においてもそうした変異が高頻度にみられることが判明するようになり,これまでの遺伝子変異と疾患との関連の証明方法に問題が見いだされるようになった.実際に,2015年の先天性QT延長症候群(cLQTS; congenital long QT syndrome)に対するデンマークの研究では,全エクソーム解析データが利用可能な870人と,遺伝子アレイデータが利用可能な6,161人について,従来cLQTSと関連するとされてきたすべての遺伝子変異を検証している.コホートで同定されたcLQTS関連8遺伝子33変異のうち,8遺伝子26変異について,QTc間隔・失神・全死亡率に影響を与えていないことが示されている28).また,2017年に報告された3世代にわたるASD大家系に対するWESによる解析においては,被疑遺伝子変異が検出されたが,機能解析においては野生型との有意差が得られず,病原性を証明できなかった29).これ以前のCHDに対するWESなどの網羅的遺伝学的解析研究について,機能解析を行うことなく原因変異が診断されているものは多く,この論文では,既存の遺伝子変異データベース上の遺伝子に,すべて病的意義があるかの疑義を指摘している.小規模の家系研究などでは,然るべき循環器関連遺伝子に,然るべき変異を認めたということだけを根拠として,機能解析など発症機序の十分な考察なく原因遺伝子変異を決定している場合もある.遺伝子変異データベースを参照する際には,変異の病原性がどのような方法で証明されているかに注意が必要である.
また,NGSの登場は膨大な数の新規遺伝子変異を見出したが,大量の遺伝子変異について病原性を解釈しなければならないという新たな課題をも生み出した.この課題に対して,米国臨床遺伝・ゲノム学会(ACMG; American College of Medical Genetics and Genomics)は,2015年に遺伝子変異の診断に関するガイドラインを示している30).本ガイドラインにおいては,メンデル遺伝性疾患において検出された変異について,様々な基準(e.g.変異頻度(MAF; Minor Allele Frequency),コンピュータ計算による機能予測であるIn silico解析,機能解析,家系情報)に基づいて,その病原性の解釈を“Pathogenic”, “Likely pathogenic”, “Uncertain significance”, “Likely benign”, “Benign”という5つに分類することを推奨している.さらに,“Likely”という分類が幅広く使用されることを見越して,当該遺伝子変異が疾患の原因である確率が90%を超えることを“Likely”の定義として推奨している.また,このガイドラインでは以下のように病原性の解釈上の注意点が記載されている.1)既存のデータベースは健常者だけではなく,疾患のある者を含んでいる可能性があること,年齢層などが偏っている可能性もあることを考慮すべきである.2)データベースに含まれる変異が最新の文献などをもとにキュレーションされアップデートされているかもチェックすべきである.3)具体的なIn silico解析ソフトウェアも挙げて(e.g. PolyPhen2, SIFT, MutationTaster),これらの有用性を紹介しているが,解析アルゴリズムの基礎は共通であるため,複数のソフトウェアを用いたとしても,これだけを根拠に病原性を決定することはできない.4)主治医に報告されたレポートについても,特に変異が“Likely pathogenic”または“Uncertain significance”であった場合には,毎年その変異についてのアップデートがないか最新情報を検索すべきである.
遺伝子変異の病的意義の評価は,NGSに限らず遺伝学的検査全般に重要である.変異があればすべて病的とは言い切れず,病原性がはっきりしない場合があること,逆に当初は病原性がはっきりしなかった変異も,その後の機能解析や症例集積の結果,病原性の有無が明確になる場合があることは知っておく必要がある.また,NGS技術が臨床応用されるにつれて,ベッドサイドでも,例えば患者説明において遺伝学の知識が要求されることが予想される.例えば意図しない「偶発的所見」は網羅的遺伝学的解析において困難な問題であり,目的とする疾患とは別にQT延長症候群や心筋症など生命のリスクにつながる遺伝子変異,発癌遺伝子などが偶発的に同定された場合に,臨床的な管理にもその家族の心理社会的にも影響が大きい31).臨床遺伝専門医のみならず,臨床に携わる小児循環器科医も,網羅的遺伝学的解析の結果を解釈するための,遺伝学の知識を習得する必要がある.
ここまでは塩基配列の変異を中心とした原因遺伝子の検索について記載してきたが,コピー数異常がCHDの原因となることも,古くから知られており,多数の先天異常症候群を含んでいる(e.g. 22q11.2欠失症候群,Williams症候群,Alagille症候群)32).Gバンド分染法などの従来の核型検査では,おおむね5 Mbより大きいコピー数異常しか検出できず,例えば22q11.2欠失症候群など微小欠失を診断するには,FISH法などの疾患特異的な手法を用いる必要があった.マイクロアレイ法は,比較的小さいコピー数異常を,DNA全長にわたって網羅的に検出できる優れた方法である.マイクロアレイ法によるCHD解析は2007年頃から報告があり,主にCHDを合併する症候群を同定するのに役立つことがわかってきている33–35).2017年に実施された多数のCHD症例に対するマイクロアレイ検査では,CHD孤発例の17.9%,知的障害や多発先天異常を合併するCHD症例ではより高い割合(20~63.2%)で,染色体の部分的なコピー数異常が検出された36).
CHDにおけるコピー数異常については,22q11.2欠失症候群やWilliams症候群など大欠失で有名なものはあるが,より小さいコピー数異常がCHDのどの程度の割合を占めるかは不明である.従来の手法であるFISH法やSanger法などでは検出が難しい,比較的小さい微小欠失・重複については,NGSが役立つ可能性がある.染色体7q11.23領域の欠失によるWilliams症候群に合併する大動脈弁上狭窄(SVAS; Supravalvular aortic stenosis)は,同領域に含まれるELN遺伝子の欠損が原因とされてきたが,このWilliams症候群の特徴を有さない,常染色体優先遺伝の家族性SVASが存在することが知られている37–40).この家族性SVASではELN遺伝子に異常を認める症例が多数報告されてきたが,一方でELN遺伝子に変異を検出できない症例が半数で見られることが知られていた41).我々の研究室では,近年NGSによるコピー数解析によって,家族性SVASでELN遺伝子に変異を検出できない症例の多くにおいて,従来のSanger法では検出が難しい,ELN遺伝子の単一あるいは複数エクソンにわたる欠失が原因であることを報告した42, 43).
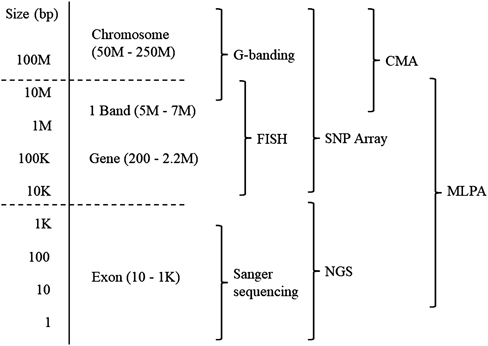
NGSは塩基配列の変異を検出するのに優れた手法であるが,コピー数解析も同時に行うことができるため,従来見過ごされてきたコピー数異常が発見される可能性がある.一方で,比較的大きいコピー数異常を定量的に検出するのには,NGSよりもマイクロアレイ法が優れており44),これらの手法は疾患や標的DNA領域のサイズによって使い分ける必要がある(Fig. 2).
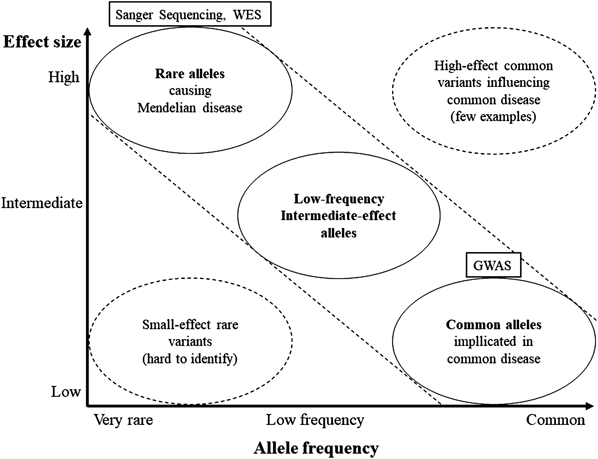
CHD大家系や症候群に伴うCHD症例に対する研究によって,数多くのCHD関連遺伝子が同定されてきたが,症例の大多数を占めるCHD孤発例の病因解明は,今後の課題である.NGSによって塩基配列異常を,またマイクロアレイ法によってコピー数異常を,それぞれ網羅的に探索することが可能となり,多くの遺伝子変異が同定されたが,殆どは家系に特異的な変異であり,CHD孤発例に応用できる普遍的な知見は乏しかった45).大家系や症候群に伴うCHD症例では,稀少かつ影響力が強い,単一遺伝子の変異が検出される場合が多いが,CHD孤発例ではこういった変異で説明可能な症例は少ない.また,CHD以外にも,自閉症や糖尿病といった様々な疾患で,遺伝子が関与していることは明らかだが,単一遺伝子の変異で説明できる疾患は限られている46).こうした疾患は,影響力が強く頻度が低い単一の遺伝子変異ではなく,影響力の弱い,よりcommonな多数の遺伝子変異の組み合わせによって,疾患発症に至るものが多いためだと考えられる47)(Fig. 3).非症候群のCHD孤発例においては,この“common disease–common variants”仮説,つまり素因となる複数の多型(SNP; Single Nucleotide Polymorphism)が存在するという仮説を証明することが次の課題となった.動物実験では,複数種のマウスにおいて,CHD関連遺伝子として有名なNkx2-5をノックアウトした研究があり,マウス種によってCHDを生じる頻度に大きな差があることから,複数の修飾遺伝子の多型が,正常な心臓発生に重要であることが示唆されている48).ある疾患にそれぞれ関与する多数のcommonな遺伝子多型の影響を解析する方法としては,遺伝的に均質な大規模コホートを用意して,多数のSNPを比較する,ゲノムワイド関連解析(GWAS; Genome Wide Association Study)が適している.2013年に中国人CHD 4,225例において,初めてGWASが実施され,1p12のTBOX転写因子に関わる遺伝子と,4q31.1にあるMAML3-Notchシグナル伝達系に関わる遺伝子に,リスクSNPを認めたと報告されている49).同年に英国などコーカシアン人種コホートでも,CHD 1,995例に対してGWASが行われ,二次孔ASDについて4q16にあるSNPに有意差を認めたものの,その他のCHDにおいては有意なSNPは見出せていないなど,CHD孤発例の原因探索においてGWASによる解析にも限界があるといえる50).
GWASはCHDの僅かな割合でしか関連のあるSNPを示せなかったが,CHD孤発例の大規模コホートに対するWES解析が普及したことで,新しい知見が報告されている.CHDの遺伝要因を探索するため,2013年に米国National Heart, Lung, and Blood Instituteは,Pediatric Cardiac Genomics Consortiumとして,10,000症例を超えるCHD大規模コホートを集積し,多数の症例にWESを実施している51).同年にZaidiらは,このCHD孤発例の大規模コホートに対して,重症CHDトリオ362組のWESデータを解析することで,初めてCHDに与えるde novo変異の影響を調査している52).解析対象を発生段階の心臓に高発現するHHE(high heart expression)遺伝子に絞ることで,これらのHHE遺伝子においてタンパク質に与える影響が大きい変異ほど有意にCHD群で多いことを示した.合計26遺伝子にde novo変異が同定されたが,その1/3がヒストン修飾遺伝子であり,エピジェネティックな制御が心臓発生に重要であることが示された.さらに,統計学的な解析を行うと,CHDの10%は,このようなde novo変異が原因であると推定された.2015年には,さらに症例数を増やしCHDトリオ1,213組のWESデータを利用して,神経発達障害を含む心外病変を伴うCHD症例は,非症候群のCHD症例と比較して,特に心臓・脳発生において高発現する遺伝子において,病原性de novo変異の影響がはるかに大きいことを示した53).この仮説は2015年に他の大規模CHDコホートでも裏付けられている54).一方でCHD孤発例では無症候の親から受け継いだ変異の影響が大きいが,同胞間でも再発率が低いことから,他のde novo変異や多因子が関わっていることが示唆された.2017年にはCHD孤発例のみを対象とした2,871組へのトリオWES解析が発表され,CHD孤発例において,de novo変異が原因の3.1%を占めることや,それまで検討されてこなかった劣性遺伝形式の変異が多く,おそらく多くのCHD関連遺伝子が未発見であることが示唆された55).また,アシュケナージ(ドイツ系ユダヤ人)において,特定の遺伝子変異が劣性遺伝することで,多数の重症CHDの原因になっていることは興味深く,遺伝的に比較的均質な本邦においても同様の知見を得られる可能性がある.2019年にはCHD 2,391症例に対するトリオWESによって,DNAH3遺伝子などの繊毛関連遺伝子や,ヒストン修飾因子などのクロマチン修飾因子関連遺伝子の変異がCHD孤発例に大きく影響していることが示された56).繊毛関連遺伝子における変異は劣性遺伝形式が多く,クロマチン修飾因子における変異はde novo変異が多く,遺伝子の機能によって遺伝形式が異なることは興味深い.繊毛関連遺伝子は内臓錯位に関連することが知られているが,同研究ではそれ以外のCHDの原因にもなっており,遺伝子の機能とその変異型が表現型に与える影響は,ネットワークのように複雑であることが示唆される.大規模コホートに対するWESは,膨大なコストや人的資源を要するが,現状で最もパワフルな研究手法であり,本邦でも同様のコンソーシアムが組織されることが期待される.
CHD孤発例の大規模コホートに対するNGS解析から,ヒストン修飾因子などのエピゲノム因子がCHD発症に大きく関わっていることが示唆されている52).心臓発生にはNKX2-5などの転写因子が深く関わっていることはよく知られており,多くのCHD症例で転写プログラムの障害が発見されてきたが,それが如何にして構造異常につながるのかは不明である.2012年にCell誌に報告された研究では,胚性幹細胞(ES細胞)を心筋細胞へ分化させる際に,経時的にどのクロマチン構造が変化しているのかを調べるゲノムワイドマッピング技術であるChiP-Seqを用いて網羅的に解析して,心臓発生に重要な制御因子を明らかにしようと試みている57).この研究ではNKX2-5など既知の転写因子の他に,心臓発生に重要な遺伝子が明らかにされ,これらの因子は,心臓発生の各段階で,複雑にエピジェネティックな制御を受けていることが判明した.2019年には,CHDに対する初のメチル化解析が報告されており,ファロー四徴の右室組織において,TBX20プロモーター領域のメチル化が低下していることが報告された58).ファロー四徴は単一遺伝子変異によるものも報告されているが,遺伝子変異が同定されない症例も多く,配列変化だけではないエピジェネティックな要因があることが示唆される.CHDは対象が心臓であるがために,生体サンプルが得がたいという困難さはあるが,エピジェネティクス解析は当該分野において発展が期待される.
小児循環器科医が,あるCHD症例に対して遺伝学的解析を考慮するのは,心外奇形を合併した症候群に伴うCHDの場合と,遺伝性の家族性CHDが疑われる場合だろう.症候群に伴うCHDの場合には,表現型から既知の疾患が疑われるなら,疾患特異的な検査法を選択すべきである.ダウン症候群やエドワーズ症候群など,染色体の数が異なる異数体が疑われた場合は,Gバンド分染法で診断できる.Williams症候群や22q11.2欠失症候群など,より小さいゲノム領域の重複または欠失が疑われる場合には,疾患特異的な検査法であるFISH法を選択する.もし表現型が22q11.2欠失症候群の場合に,欠失領域が小さく,既存のFISH法で検出できない場合には,ほとんどの疾患において保険未収載の検査だが,MLPA法を行うことで検出できる可能性がある59).Noonan症候群におけるPTPN11遺伝子など,特定の単一遺伝子変異による症候群が疑われる場合には,Sanger法を用いて変異を同定する.表現型から鑑別診断が絞れない場合には,保険未収載であり,研究の一環としての手法だが,NGS解析やマイクロアレイ法を考慮する.
遺伝性の家族性CHDの場合には,単一遺伝子の塩基変異が原因となる可能性が高いが,表現型から原因遺伝子を想定することはほとんどの場合に困難なため,コストが許容できるのであれば,NGSによる全エクソーム解析または疾患遺伝子パネル解析が第一選択である.発端者に対するNGS解析で,病的な変異が同定されなかった場合には,さらに血縁の発症者など解析対象を増やすことで,原因変異を絞ることができる場合がある.また,NGSを用いた全ゲノムまたは全エクソーム解析では,同じ配列が検出された回数(リード数)を元にコピー数を推定することも可能である.FISH法などの既存の検査法で検出できないような小さいサイズの欠失・重複については,NGSによるコピー数解析が役立つ場合もある.一方で,NGSによるコピー数解析の定量性は高くない.症候群に伴うCHDは,塩基配列の変異よりもコピー数の異常が原因である割合が多いため,NGSで原因が特定できない場合に,マイクロアレイ法などの定量性に優れた網羅的解析法を組み合わせることで,原因が特定できる可能性が高まる31).これらの解析手法は,検出可能な標的DNA領域のサイズや種類が異なるため,疾患に応じて適切な手法を選択することが重要である.
原因遺伝子が判明することは,CHD診療において,遺伝カウンセリングや予後・合併症の予測に寄与する.例えば既知の症候群に伴うCHDであることが判明した場合に,心外先天異常などの合併症に対して早期に対応することが可能となる16).単一遺伝子異常に伴うCHDでも,遺伝学的解析が,管理や治療法選択の参考になる場合がある.例えば,NKX2-5遺伝子異常に伴うCHDは,致死的な伝導障害を合併することが報告されており9),定期的に心電図を確認することで予期せぬ突然死を防げる可能性がある.NGS解析などの網羅的遺伝学的解析手法が臨床応用され,普及することで,症例データが集積し,臨床に応用できる知見が積み上がっていくことが期待される.
NGSによって得られる膨大な遺伝子変異リストの中から原因遺伝子を見つけ出すことの困難さは,“a needle in haystack”と例えられるが25),ACMGによる病原性判定のガイドライン策定や,大規模コホートへの解析によるデータベースの整備などによって,徐々に解析環境は改善している.遺伝子変異データベースを共有することで,膨大な健常コントロールを用意する必要がなくなるだけではなく,稀少な疾患について効率的に症例を集積することが可能となった.また,新世代のNGSでは従来の数百塩基を超えるロングリード(長い塩基対の解読)が可能となり,繰り返し配列や長い欠失など,従来は困難な配列の解読も可能となった.更にNGS解析自体のコストも加速度的に下がっており,パネルシーケンス時代からWES解析の時代を経て,イントロン領域も解析可能な全ゲノム解析の時代も遠くないと思われる.大規模コホート解析と,良質な機能解析を伴う稀少な症候群や家系の解析が,両輪となってCHDに対する遺伝子研究を発展させることが期待される.
謝辞Acknowledgments
本稿執筆にあたり有益な助言を頂いた,名古屋大学大学院医学系研究科成長発達医学の加藤太一准教授に深謝する.
引用文献References
1) Taussig HB: Evolutionary origin of cardiac malformations. J Am Coll Cardiol 1988; 12: 1079–1086
2) Bentham J, Bhattacharya S: Genetic mechanisms controlling cardiovascular development. Ann NY Acad Sci 2008; 1123: 10–19
3) Ferencz C, Rubin JD, McCarter RJ, et al: Congenital heart disease: Prevalence at livebirth. The Baltimore-Washington Infant Study. Am J Epidemiol 1985; 121: 31–36
4) Nees SN, Chung WK: The genetics of isolated congenital heart disease. Am J Med Genet C Semin Med Genet 2020; 184: 97–106
5) Wang X, Wang J, Zhao P, et al: Familial congenital heart disease: Data collection and preliminary analysis. Cardiol Young 2013; 23: 394–399
6) Chen CM, Bentham J, Cosgrove C, et al: Functional significance of SRJ domain mutations in CITED2. PLoS One 2012; 7: e46256
7) Basson CT, Bachinsky DR, Lin RC, et al: Mutations in human TBX5 cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet 1997; 15: 30–35
8) Mori AD, Bruneau BG: TBX5 mutations and congenital heart disease: Holt-Oram syndrome revealed. Curr Opin Cardiol 2004; 19: 211–215
9) Schott JJ, Benson DW, Basson CT, et al: Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 1998; 281: 108–111
10) Arrington CB, Bleyl SB, Matsunami N, et al: Exome analysis of a family with pleiotropic congenital heart disease. Circ Cardiovasc Genet 2012; 5: 175–182
11) Bamshad MJ, Ng SB, Bigham AW, et al: Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 2011; 12: 745–755
12) Abecasis GR, Auton A, Brooks LD, et al: Genomes Project C: An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491: 56–65
13) Al Turki S, Manickaraj AK, Mercer CL, et al: UK10K Consortium: Rare variants in NR2F2 cause congenital heart defects in humans. Am J Hum Genet 2014; 94: 574–585
14) Greenway SC, McLeod R, Hume S, et al: FORGE Canada Consortium: Exome sequencing identifies a novel variant in ACTC1 associated with familial atrial septal defect. Can J Cardiol 2014; 30: 181–187
15) D’Alessandro LC, Al Turki S, Manickaraj AK, et al: Exome sequencing identifies rare variants in multiple genes in atrioventricular septal defect. Genet Med 2016; 18: 189–198
16) Blue GM, Kirk EP, Giannoulatou E, et al: Targeted next-generation sequencing identifies pathogenic variants in familial congenital heart disease. J Am Coll Cardiol 2014; 64: 2498–2506
17) Jia Y, Louw JJ, Breckpot J, et al: The diagnostic value of next generation sequencing in familial nonsyndromic congenital heart defects. Am J Med Genet A 2015; 167A: 1822–1829
18) Pulignani S, Vecoli C, Borghini A, et al: Targeted next-generation sequencing in patients with non-syndromic congenital heart disease. Pediatr Cardiol 2018; 39: 682–689
19) van Nimwegen KJ, van Soest RA, Veltman JA, et al: Is the $1000 genome as near as we think? A cost analysis of next-generation sequencing. Clin Chem 2016; 62: 1458–1464
20) Szot JO, Cuny H, Blue GM, et al: A screening approach to identify clinically actionable variants causing congenital heart disease in exome data. Circ Genom Precis Med 2018; 11: e001978
21) Wang X, Charng WL, Chen CA, et al: Germline mutations in ABL1 cause an autosomal dominant syndrome characterized by congenital heart defects and skeletal malformations. Nat Genet 2017; 49: 613–617
22) Yamamoto H, Hayano S, Okuno Y, et al: Phosphorylated proteome analysis of a novel germline ABL1 mutation causing an autosomal dominant syndrome with ventricular septal defect. Int J Cardiol 2021; 326: 81–87
23) Andersen TA, Troelsen Kde L, Larsen LA: Of mice and men: Molecular genetics of congenital heart disease. Cell Mol Life Sci 2014; 71: 1327–1352
24) Paige SL, Saha P, Priest JR: Beyond gene panels: Whole exome sequencing for diagnosis of congenital heart disease. Circ Genom Precis Med 2018; 11: e002097
25) Cooper GM, Shendure J: Needles in stacks of needles: Finding disease-causal variants in a wealth of genomic data. Nat Rev Genet 2011; 12: 628–640
26) Jiang JQ, Li RG, Wang J, et al: Prevalence and spectrum of GATA5 mutations associated with congenital heart disease. Int J Cardiol 2013; 165: 570–573
27) Kodo K, Nishizawa T, Furutani M, et al: Genetic analysis of essential cardiac transcription factors in 256 patients with non-syndromic congenital heart defects. Circ J 2012; 76: 1703–1711
28) Ghouse J, Have CT, Weeke P, et al: Rare genetic variants previously associated with congenital forms of long QT syndrome have little or no effect on the QT interval. Eur Heart J 2015; 36: 2523–2529
29) Blue GM, Humphreys D, Szot J, et al: The promises and challenges of exome sequencing in familial, non-syndromic congenital heart disease. Int J Cardiol 2017; 230: 155–163
30) Richards S, Aziz N, Bale S, et al: ACMG Laboratory Quality Assurance Committee: Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424
31) Andelfinger G: Next-generation sequencing in congenital heart disease: Do new brooms sweep clean? J Am Coll Cardiol 2014; 64: 2507–2509
32) 上砂光裕:ここまで知っておきたい発生学—先天性心疾患の遺伝子解析—.日小児循環器会誌2018; 34: 105–110
33) Dorn C, Grunert M, Sperling SR: Application of high-throughput sequencing for studying genomic variations in congenital heart disease. Brief Funct Genomics 2014; 13: 51–65
34) Sanchez-Castro M, Gordon CT, Petit F, et al: Congenital heart defects in patients with deletions upstream of SOX9. Hum Mutat 2013; 34: 1628–1631
35) Thienpont B, Mertens L, de Ravel T, et al: Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur Heart J 2007; 28: 2778–2784
36) Wu XL, Li R, Fu F, et al: Chromosome microarray analysis in the investigation of children with congenital heart disease. BMC Pediatr 2017; 17: 117
37) Beuren AJ, Schulze C, Eberle P, et al: The syndrome of supravalvular aortic stenosis, peripheral pulmonary stenosis, mental retardation and similar facial appearance. Am J Cardiol 1964; 13: 471–483
38) Eisenberg R, Young D, Jacobson B, et al: Familial supravalvular aortic stenosis. Am J Dis Child 1964; 108: 341–347
39) Williams JC, Barratt-Boyes BG, Lowe JB: Supravalvular aortic stenosis. Circulation 1961; 24: 1311–1318
40) Merla G, Brunetti-Pierri N, Piccolo P, et al: Supravalvular aortic stenosis: Elastin arteriopathy. Circ Cardiovasc Genet 2012; 5: 692–696
41) Metcalfe K, Rucka AK, Smoot L, et al: Elastin: Mutational spectrum in supravalvular aortic stenosis. Eur J Hum Genet 2000; 8: 955–963
42) Hayano S, Okuno Y, Tsutsumi M, et al: Frequent intragenic microdeletions of elastin in familial supravalvular aortic stenosis. Int J Cardiol 2019; 274: 290–295
43) Hayano S, Okuno Y, Tsutsumi M, et al: Corrigendum to “Frequent intragenic microdeletions of elastin in familial supravalvular aortic stenosis.” Int J Cardiol 2019; 292: 283
44) 野村文夫:遺伝学的検査,福嶋義光(編): 遺伝医学やさしい系統講義18講 first ed. 東京,メディカルサイエンスインターナショナル,2013, pp177–194
45) Blue GM, Kirk EP, Giannoulatou E, et al: Advances in the genetics of congenital heart disease: A clinician’s guide. J Am Coll Cardiol 2017; 69: 859–870
46) Maher B: Personal genomes: The case of the missing heritability. Nature 2008; 456: 18–21
47) Manolio TA, Collins FS, Cox NJ, et al: Finding the missing heritability of complex diseases. Nature 2009; 461: 747–753
48) Winston JB, Erlich JM, Green CA, et al: Heterogeneity of genetic modifiers ensures normal cardiac development. Circulation 2010; 121: 1313–1321
49) Hu Z, Shi Y, Mo X, et al: A genome-wide association study identifies two risk loci for congenital heart malformations in Han Chinese populations. Nat Genet 2013; 45: 818–821
50) Cordell HJ, Bentham J, Topf A, et al: Genome-wide association study of multiple congenital heart disease phenotypes identifies a susceptibility locus for atrial septal defect at chromosome 4p16. Nat Genet 2013; 45: 822–824
51) Gelb B, Brueckner M, Chung W, et al: Pediatric Cardiac Genomics Consortium: The congenital heart disease genetic network study: Rationale, design, and early results. Circ Res 2013; 112: 698–706
52) Zaidi S, Choi M, Wakimoto H, et al: De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013; 498: 220–223
53) Homsy J, Zaidi S, Shen Y, et al: De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015; 350: 1262–1266
54) Sifrim A, Hitz MP, Wilsdon A, et al: INTERVAL Study; UK10K Consortium; Deciphering Developmental Disorders Study: Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet 2016; 48: 1060–1065
55) Jin SC, Homsy J, Zaidi S, et al: Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet 2017; 49: 1593–1601
56) Watkins WS, Hernandez EJ, Wesolowski S, et al: De novo and recessive forms of congenital heart disease have distinct genetic and phenotypic landscapes. Nat Commun 2019; 10: 4722
57) Paige SL, Thomas S, Stoick-Cooper CL, et al: A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell 2012; 151: 221–232
58) Gong J, Sheng W, Ma D, et al: DNA methylation status of TBX20 in patients with tetralogy of Fallot. BMC Med Genomics 2019; 12: 75
59) Stachon AC, Baskin B, Smith AC, et al: Molecular diagnosis of 22q11.2 deletion and duplication by multiplex ligation dependent probe amplification. Am J Med Genet A 2007; 143a: 2924–2930