小児期発症の特発性拘束型心筋症Idiopathic Restrictive Cardiomyopathy in Children
大阪大学大学院医学系研究科小児科学Department of Pediatrics, Osaka University Graduate School of Medicine ◇ Osaka, Japan
発行日:2021年11月1日Published: November 1, 2021
拘束型心筋症(restrictive cardiomyopathy; RCM)は,心室拡張障害を主体とし,心室壁厚や心収縮が概ね保たれている心筋症と定義され,小児心筋症の中でも稀な疾患である.小児期発症の特発性RCMは非常に予後が悪く,診断後2年の心臓移植/死亡回避率は約40%と報告されている.また,進行する心室拡張障害に対するエビデンスのある治療法は存在せず,最終的に心臓移植でしか救命できない症例が多い.本稿では,小児RCMの疾患概念から疫学,予後,臨床検査,治療について概説し,加えて我々の施設での小児期発症RCM症例の臨床像を提示する.また,近年明らかになってきているRCMに関連する遺伝子異常について,さらに,我々の行っているRCMに対する基礎研究についても報告する.
Restrictive cardiomyopathy (RCM) is characterized by impaired ventricular diastolic function with preserved ventricular contraction and wall thickness. The prognosis of young children with idiopathic RCM is poor, and the 2-year transplant-free survival rate has been reported to be approximately 40%. No medical treatment has been established for ventricular diastolic dysfunction; thus, heart transplantation is the only way to cure end-stage heart failure in patients with RCM. Herein, the pathology, etiology, clinical examinations, and medical management of pediatric patients with RCM were reviewed. This article includes a clinical case review of pediatric patients with RCM in our institute and the results of in vitro experiments using cardiac fibroblasts derived from patients with RCM and exome sequencing.
Key words: restrictive cardiomyopathy; pediatric; heart transplantation; ventricular assist device; exome sequence
© 2021 特定非営利活動法人日本小児循環器学会© 2021 Japanese Society of Pediatric Cardiology and Cardiac Surgery
拘束型心筋症(restrictive cardiomyopathy; RCM)は,心室拡張不全を主体とする心筋症で,心収縮は概ね保たれており心室拡大や心室壁肥厚を通常認めないものと定義される.小児期発症のRCMは,小児心筋症の2~5%を占めるとされ,小児人口10万人当たりの発症率は0.3人と推定される,かなり稀な疾患である.小児では,心アミロイドーシスや心サルコイドーシス,虚血性心疾患などに伴う二次性の拘束型心筋症の頻度は少なく,多くが特発性/遺伝性RCMであるが,心室の拡張障害を主体とする疾患のうち,収縮性心膜炎や心内膜弾性線維症との鑑別は必要である.
1998年から2008年の北米における小児心筋症レジストリデータの解析によると,RCMと診断された患者は152名で,平均診断年齢は6.2歳,男性は48%,家族歴を有する者が23%であった1).小児RCMの予後は,拡張型心筋症(dilated cardiomyopathy; DCM)や肥大型心筋症(hypertrophic cardiomyopathy; HCM)などほかの心筋症に比べて非常に悪く,診断から2年の心臓死回避率(心臓移植および死亡の回避率)は40%とされている.さらに,同論文では小児RCMにおける2つのサブグループ解析が行われており,1つは典型的なRCMともいえる,両心房の著明な拡大を有するが心室壁肥厚を呈さないpure RCM,もう1つは心室壁の肥厚を認めるが拡張障害が病態の主体となるHCM/RCM overlapと呼ばれる病態グループである.この2つのサブグループに分類した場合,pure RCMが特に予後が悪いと報告されており,診断後2年と5年の心臓死回避率はそれぞれ,34% vs 53%と,22% vs 43%(=pure RCM vs HCM/RCM)であった.2018年に発表されたMayo Clinicからの報告では,1975年から2013年に診断された小児RCMは20例で,その5年心臓死回避率は38%であり,1994年から2013年の症例と1975年から1993年の症例とでは,予後に大きな差を認めなかったとされている2).これら北米からの報告は我々の施設での経験と大きく変わりなく,当院でのRCMの臨床像については後述したい.
RCMの基本病態は,心室stiffnessの増大による心室コンプライアンスの低下である.それにより,心室の充満にはより高い圧が必要となり,左右心房圧の上昇と心房容積の拡大が見られるようになる.病初期には心収縮は保たれており,心拍出量も維持されているため,臨床症状はわかりにくい.単に運動が得意ではない子,すぐ疲れたと訴える子,とだけ認識されているケースがほとんどである.わが国では,学校心臓検診の心電図検査において,心房負荷所見やST変化,T波異常で検知される場合がある.胸部レントゲンでは初期は所見に乏しく診断は困難であるが,心房拡大所見,肺うっ血所見が認められれば診断の契機になる.自覚症状が乏しい場合は,学校心臓検診における心電図異常か,偶然撮影された胸部X線写真における心拡大から,最終的に心エコー検査を施行することによって,RCMの診断に至ることが多い.
病期が進行すると,心収縮も次第に低下し,心室容積が小さくなることや心拍数が低下することもあって,心拍出量が低下して労作時倦怠感,運動不耐が顕在化する.ただ我々の経験では,労作時倦怠感だけで積極的にRCMが診断されることは少ない.また,低心拍出症状は,突発的でありかつ一過性のことがしばしばある.すなわち,「普段は元気で特に症状は認められないが,時折顔色不良と腹痛,気分不良を訴える.しばらく休むと回復してまた元気になる」という症状が典型的である.しかしこのような症状は非特異的であり,問診で得られる自覚および他覚症状のみでRCMを疑うことは困難である.病態が進行すると,症例によっては左室の拡張障害が優位なケースと右室の拡張障害が優位なケースがあり,前者では肺うっ血に伴う症状が主体となり,咳嗽,喘鳴や喀血を主訴とする場合がある.後者では,肝うっ血などの腹部臓器うっ血が主体となり,そのようなケースでは自覚症状が出ることはまれであるが,肝腫大や浮腫からRCMの診断に至るケースがある.また,病態が進行するにつれて徐脈,心室性不整脈を認める頻度が増加することが多い.終末期には心室収縮がさらに低下し,低心拍出に伴う心不全症状に加えて,肺うっ血による呼吸不全,肝うっ血,腎うっ血,腸管うっ血による多臓器不全が進行する.また,致死性不整脈による死亡リスクが上昇する.
病初期では明らかな所見を認めない場合がある.心房拡大の進行とともに,右第2弓,左第3弓の突出を認め,肺うっ血所見を認める(Fig. 1A).
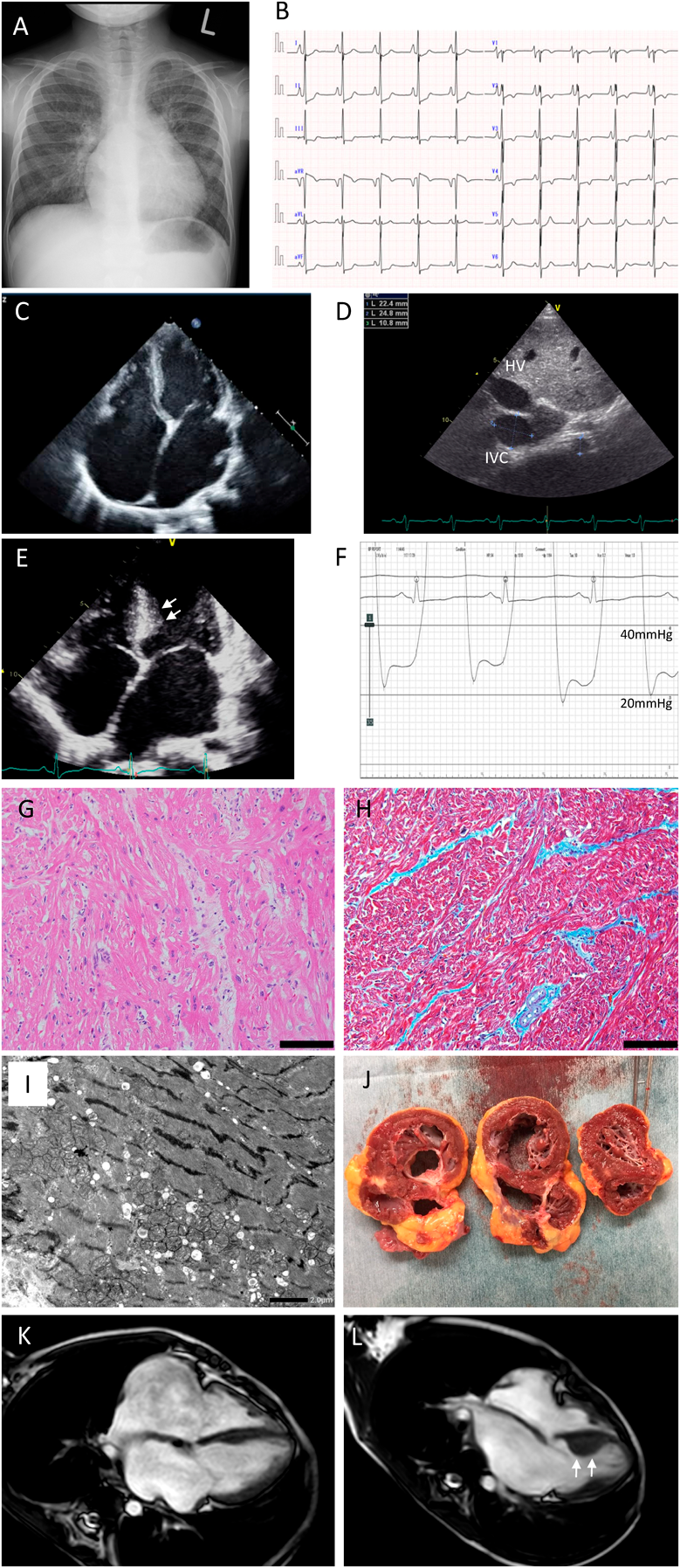
(A) Chest X-ray of 6-years old pure RCM patient. Note the severe pulmonary congestion. (B) Electrocardiogram of 7-years old pure RCM patient. (C) Echocardiogram of four chamber view. Both right and left atrium are extremely dilated with normal ventricular size. (D) Inferior vena cava (IVC) as well as hepatic vein (HV) are significantly dilated. (E) Echocardiogram four chamber view of 7-years old HCM/RCM patient. The arrows show asymmetrical septal hypertrophy. (F) Pressure curve of left ventricle, showing typical dip and plateau pattern and elevation of end diastolic pressure. (G) Hematoxylin–Eosin stain of 11-years old pure RCM patient. Some of myocytes show hypertrophy and disarray. Bar is 100 µm. (H) Masson-Trichrome stain of the same patient. There is mild interstitial fibrosis (blue stain). Bar is 100 µm. (I) Electron microscope image of the same patient. Bar is 2 µm. (J) Macroscopic finding of the same patient. (K) Cardiac magnetic resonance image of 6-years old pure RCM patient. (L) Cardiac magnetic resonance image of 7-years old HCM/RCM patient, same as panel E. The arrows show asymmetrical septal hypertrophy. RCM, restrictive cardiomyopathy.
病初期には最も早く異常所見が出やすく診断の契機となることがある.心房負荷所見として,先鋭P波や二相性P波を認める.右肩上がりに上昇したSTに続き,ノッチ付きまたは二相性のピークが遅いT波が出現する特徴的な所見を認めることもあるとされる3).また,陰性T波を伴うST低下を伴うことがある(Fig. 1B).病期の進行とともに,洞性徐脈の進行や心室性不整脈の出現が見られる場合がある.最近のMurajiらの報告では,V1誘導における二相性P波の陽性成分と陰性成分の電位の合計が小児RCMにおいて有意に高値であり,特に426 µVを超える場合に,感度71%,特異度89%で,心移植・補助人工心臓装着・死亡のリスクを予測できるとしている4).
まず特徴的な所見として両心房の拡大を認める(Fig. 1C).駆出率はほぼ正常であることが多い.病期の進行とともに拡張障害の指標となる房室弁流入血のE/Aが上昇し,E波減速時間の短縮,E/e’の上昇が認められるとされるが,小児RCMでは心エコーで一般的に用いられる拡張障害指標は明らかでないケースもしばしばある5).三尖弁輪収縮期移動距離や僧帽弁輪収縮期移動距離は心拍出量が保たれている状態では正常であることが多い.左室拡張障害の進行による左房圧の上昇とともに肺高血圧が顕在化しているケースでは,三尖弁逆流速度の上昇が認められる.また,右室拡張障害を反映して,右房拡大に加えて下大静脈と肝静脈の拡張や逆流,呼吸性変動の消失が認められる(Fig. 1D).一部の症例では心室中隔の非対称性肥厚を認め,このような症例ではHCM/RCM overlapとして診断される(Fig. 1E).
ほとんどの症例で,BNP(b-type natriuretic peptide),NT-proBNP値の上昇が認められる.特に値の経時的推移が治療方針の大きな参考所見になる.右室拡張障害により肝うっ血が進行しているケースでは,主にγGTPが上昇している.T-BILも軽度上昇している場合がある.トランスアミナーゼは上昇していないことがほとんどである.
上記の検査でRCMを強く疑う場合には必須の検査である.初診時には,両心室の拡張末期圧の上昇,中心静脈圧の上昇,肺動脈楔入圧の上昇が認められる.特徴的には左室圧曲線はdip and plateauを描き拡張末期圧の上昇を認める(Fig. 1F).左室拡張障害が強いケースでは,左房圧上昇に伴う肺高血圧(Group 2の肺高血圧)を認める.病初期では,肺血管抵抗係数(pulmonary vascular resistance index; PVRI)は正常範囲であることが多いが,進行したケースでは肺血管リモデリングの進行とともにPVRIも上昇しており,いわゆるcombined pulmonary hypertensionを呈していることがある.PVRIが9.0 Wood単位・m2を超えるケースでは,もはや心臓移植適応から外れ心肺同時移植を考えなければならないため,心臓移植適応であるかどうかの検討には,急性血管反応試験が必要になる.一部の特異な例を除くほとんどの小児RCMでは肺血管反応性はまだ保たれていることが多いが,酸素負荷試験,一酸化窒素負荷試験は,RCM患者では重度の肺うっ血を惹起するため実施には細心の注意が必要である.可能な限り短時間での検査に留め,十分に経験のある施設で行い,心臓外科医や集中治療医に事前に連絡したうえで,体外循環などのバックアップ体制を整えて実施したほうがよい.心室造影検査は,心エコーや心臓MRIなどほかのモダリティでも検査できる場合には必須ではなく,拡張末期圧が著しく高いケースでは避けたほうがよい場合もある.
上記の心臓カテーテル検査と同時に,心内膜心筋生検も実施することが望ましい.特に小児のRCMでは心室内腔が大きくなく,適切な位置に生検鉗子を導くのが困難なケースもある.そのようなケースでは大腿静脈アプローチよりは内頸静脈アプローチが有用である.特発性RCMの場合,心筋生検では特異的な所見は少ない.軽度から中等度の間質線維化や心筋細胞の肥大,配列の乱れ,核の大小不同等が所見として認められる(Fig. 1G, 1H).電子顕微鏡検査も特異的な所見は少ない.異常蓄積物の有無やミトコンドリア形態など,二次性心筋症の鑑別が主な目的となる(Fig. 1I).心臓移植の際に得られた摘出心を観察すると,心内膜線維化は軽微な症例が多い.心室壁厚は正常範囲かやや厚めであり,心室壁内のところどころに白色の肉眼的線維化を認める部位が存在する場合がある(Fig. 1J).
形態的には拡大した両心房と正常サイズあるいは小さくなった心室を認める(Fig. 1K).病期の進行とともに,心室拡張容積が低下することが多い.HCM/RCM症例では,心エコー所見と同様に肥厚した左室壁を確認することができる(Fig. 1L).心収縮はほとんど保たれているか軽度低下している.ガドリニウム造影では,遅延造影陽性所見を認めることはほとんどない.T1-mappingでは病期の進行に伴って,native T1値の上昇やextra cellular volume(ECV)の上昇を認めるケースが多い.特にECVは心筋生検で観察される間質線維化の程度とよく相関している(Ishii et al. unpublished data).
近年,RCMの原因遺伝子としていくつかの遺伝子バリアントが報告されている.主として,トロポニンやミオシンなど心筋細胞構造タンパクであり,TNNI3,MYL2,MYH7,TNNT2などが報告されている6-8).しかし,次世代シークエンサーを用いた全エクソン解析においても,有意な遺伝子バリアントが見つかる症例は約半分程度と報告されている.わが国における報告では,Hayashiらが7例中5例(71%)と報告している9).下述するように我々の施設における全エクソン解析においては,18例中10例(56%)で病原性があると判断されるバリアントを同定でき,ほとんどがTNNI3のミスセンスバリアントであった.
心室拡張障害を主体とするRCMに対して,有効性の証明されたエビデンスのある管理法・治療法は存在しない.
心室性不整脈リスクの高いRCMでは,一定以上の運動制限は必要と考えられる.不整脈の種類や頻度,自覚症状に応じて,学校生活管理区分はB~Dでの管理が望ましい.また,ある程度の水分・塩分管理も望ましいが,厳しい水分制限は病態の進行とともに低心拍出症状を増悪させるため,患者の自覚症状や理学所見,各種検査所見に合わせてケースバイケースで考えなければならず,病態の進行とともに修正していく必要性がある.
診断初期で心拍出量の維持されているケースでは,うっ血改善のため利尿薬が投与される.furosemideに加えて,抗アルドステロン薬としてspironolactoneが投与されることがある.それらで効果が乏しい場合は,tolvaptanを追加することも考慮されるが小児では適応外となる.病期の進行とともに低心拍出症状(突発的な顔色不良や腹痛,嘔吐など)の頻度が増加してくることが多いため,利尿薬は減量を余儀なくされる場合がある.進行期には,臓器うっ血と低心拍出症状のバランスをとることが困難な状況へと追い込まれることが多い.
心室後負荷の低減,リモデリング抑制など抗心不全効果を期待して投与される.しかしこれも,病期の進行とともに低心拍出症状や低血圧との兼ね合いから積極的な増量は難しいケースがある.
RCMに対するエビデンスはなく,また,徐脈のケースも多いため積極的に導入はしにくい.比較的心拍数の高いケースでは,心室拡張時間の延長を期待して導入される場合もありうるが,その可否や適切な投与量の判断は難しく,個々の症例に応じて調整せざるを得ない.
心房拡大が著明なRCMでは,心房内血栓の予防として抗血小板療法あるいは抗凝固療法を考慮したほうがよいと考えられる.
心室頻拍が認められる症例では,amiodaroneの導入が考慮される.心室頻拍や心室細動による失神の既往のある症例では,二次予防として植込み型除細動器の導入が考慮される.病態の進行とともに徐脈が進行し,心拍出量を維持できないケースでは,ペースメーカ植込みが考慮される.
上記のように,小児RCMは予後が非常に悪い疾患であるとともに,有効な内科的治療法が存在しないことから,診断された時点から将来的な心臓移植について検討し,本人および家族に情報提供されるべき疾患である.ただし,一部のRCM,特にHCM/RCMでは緩徐に病態が進行して,長期に内科管理が可能な症例が一定の割合で存在する.
心臓移植は特殊な医療である.それは通常の薬物治療や外科手術などと異なり,ドナーとその遺族の善意と崇高なる願いによって成り立っている医療だからである.したがって,心臓移植適応については公正かつ厳正に判断され,患者の心臓の状態,心外臓器の状態といった純粋な医学的評価ともに,患者本人と家族全員の移植医療への理解,意識やコンプライアンス,社会的・経済的サポート体制が適応判定委員会において検討される.そして,移植医療は決して治療のゴールではなく,そこから様々な移植後合併症との闘いが新たにスタートする.これまでのわが国における心臓移植後の成績は世界のそれと比較しても非常に良好で,10年生存率は約90%であるが,これはドナーの残した想いに応えようとするレシピエントとその家族の高い意識と医学管理の賜物であると考えられる.しかし,わが国において依然ドナー数は少なく,移植までの待機期間は長い.特に小児RCMは,病初期にはほとんど症状がないにもかかわらず,一旦進行し始めると有効な治療法がないだけに,数年単位に及ぶ待機期間を見据えながら,安全に移植医療に到達する道を考えなければならず,Status 1での移植待機に入る至適時期の判断が難しい.
さらに,RCMでは補助人工心臓管理も非常に困難である.その理由の一つとして心室が拡大しておらずむしろ小さいこと,さらには,左室だけでなく右室不全も合併しているため,左室補助装置(left ventricular assist device; LVAD)によるサポートのみでは右心不全の管理が困難であることが挙げられる.我々の施設での経験では,pure RCMに対するLVAD装着の症例では,ほとんどの症例で移植待機中のどこかの時点からカテコラミン持続静注の追加による右室サポートをはかる必要があり,それでも心不全管理の困難なケースでは,RVAD(right ventricular assist device)を追加して,両室補助(biventricular assist device; BiVAD)を選択せざるを得ない場合がある10)(Fig. 2).しかし,RCMにおけるBiVADは循環補助の管理がさらに困難であり,体血管抵抗や肺血管抵抗,LVAD, RVADのわずかな駆出バランスの変化によって,一気に肺うっ血を来たしたり,脱血不良,低心拍出症状を来たす.これまでも,BiVAD管理はLVAD管理と比較して死亡率が高いことが報告されている11).
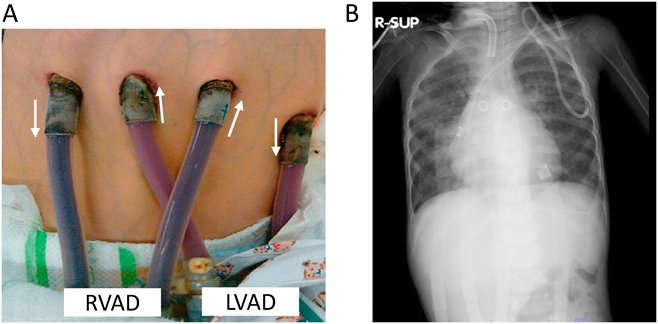
(A) The image of cannulation of EXCOR Pediatric BiVAD. (B) Chest X-ray image when the patient suffered from pulmonary congestion with BiVAD. BiVAD, biventricular assist device; LVAD, left ventricular assist device; RCM, restrictive cardiomyopathy; RVAD, right ventricular assist device.
まとめると,RCMでは補助人工心臓導入に至る前に移植に到達することが理想的と考えられるが,我が国の移植待機期間を考慮すると,数年先の病状を見積もって現時点での治療方針を決定する,という医学判断は現実的には非常に難しい.心臓移植実施施設と早めに連携を行いながら,各症例の条件に応じて検討していかざるを得ないと考えられる.
先述したように,北米におけるレジストリ研究や,米国センター病院の経験においても小児RCMの症例数は少ない1, 2, 12).当院では,1998年から2020年までの間に33例の小児RCM(pure RCM 23例,HCM/RCM 10例)を経験しており,その臨床像について報告する.
性別は女性17例,男性16例,平均診断時年齢は6.3±4.0歳であった.pure RCMの平均診断時年齢は5.1±3.7歳,HCM/RCMの平均診断時年齢は9.0±3.7歳であり,pure RCMのほうが有意に若年であった(Mann–Whitney U test:p=0.037)(Fig. 3A).非持続性を含む心室頻拍を認めた症例は全体で6例(18%)であった.pure RCMの5例(15%)で心臓移植待機中にVAD植込みを行っており,うち1例ではBiVADが導入された.また,pure RCM 23例中22例(96%)が心臓移植もしくは移植待機中であったのに対して,HCM/RCMでは移植もしくは待機中なのは10例中5例(50%)であった(Fig. 3B).すなわち北米での研究結果と同じく,pure RCMはHCM/RCMに比してさらに予後が悪いことが推測された.そこで,心臓死回避率(survival rate free from transplant/death)についてカプランマイヤー曲線による解析を行った.診断後2年と5年の心臓死回避率はそれぞれ,30% vs 100%と,20% vs 70%(=pure RCM vs HCM/RCM)であった.log-rank testにて2群間に有意差(p=0.0003)を認め,既報のとおりpure RCMはHCM/RCMより予後不良であることが明らかとなった(Fig. 3C).我が国の小児RCMの臨床像として,興味深いのは診断の契機であった.学校心臓検診における心電図異常を契機として診断に至った症例がRCM全体で18例(55%)あり,うち小学1年が12例,中学1年が6例であった.欧米とは異なる我が国独自のシステムとして,学校心臓検診の重要性がRCMの診断においても明らかであった.その他の診断契機としては,感染症罹患時に撮影された胸部X線写真における心拡大が3例(By chance XP),うっ血症状(浮腫や肝腫大など)が6例(Edema),低心拍出症状(倦怠感や顔色不良など)が3例(LOS),ほか,心雑音,家族歴,徐脈,が1例ずつ(Others)であった.pure RCMのほうが,臨床症状出現によって診断に至った症例が多い傾向にあった(Fig. 3D).しかし,これらの解析では,当施設が小児心臓移植実施施設であるため,より重症例が集積しやすいという患者選択バイアスがあり,それが本解析のlimitationとして考えられた.
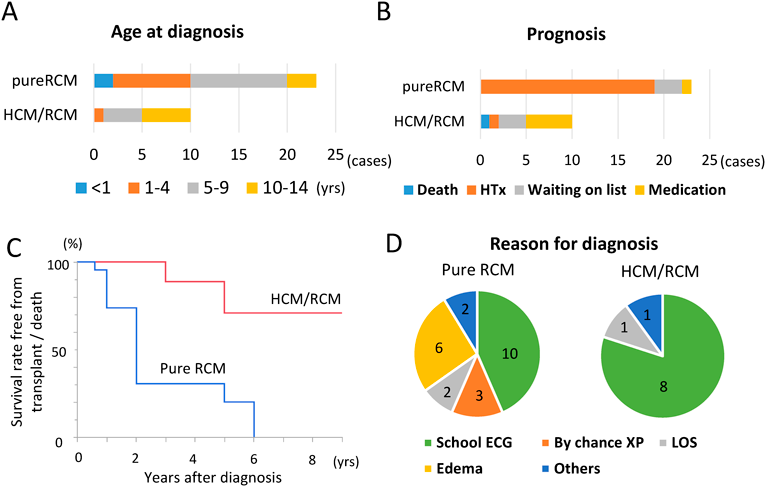
(A) Age distribution when the patients were diagnosed as RCM. (B) The prognosis of pediatric RCM patients. (C) The Caplan-Meier curve for the survival rate free from transplant and death in pure RCM and HCM/RCM patients. (D) Reason for diagnosis as RCM. HCM, hypertrophic cardiomyopathy; HTx, heart transplantation; LOS, low output syndrome; RCM, restrictive cardiomyopathy.
全エクソン解析の結果が現時点で判明している18例中,病原性があることがほぼ確実と思われる(アレル頻度が0.5%未満で,複数の機能予測プログラムで障害性が予測されており,かつ,類似のRCMもしくはHCM表現型を呈する複数の既報がある)バリアントを同定できた症例は10例(56%)で,TNNI3が8例,その他MYH7とMYL2のミスセンスバリアントであった.
これまで述べたpure RCMやHCM/RCMといったサブタイプのほかにも,非常に特異な臨床像を示し,著明な拡張障害を来す心筋症を我々は経験している.それは,左室心尖部を欠く球状の小さな左室を呈する拘束型心筋症で,この左室形態はisolated left ventricular apical hypoplasiaとしていくつかの症例報告が散見される13–15).しかし,ほとんどの症例は軽度の心不全症状や肺うっ血を呈する程度であるとされ,時に致死性不整脈による突然死が報告されている.一方,我々の経験した3症例は,著明な左室拡張末期圧の上昇だけでなく,体血圧を超える著明な肺高血圧,20 Wood単位・m2を超える肺血管抵抗係数を示し,1例は死亡,2例は心肺同時移植に至った(Fig. 4).わが国で施行された心肺同時移植3例のうち2例はこの疾患であった.これらの症例に対する全エクソン解析を行ったが,症例に共通する有意なバリアントを認めておらず,現在も解析を続けている.
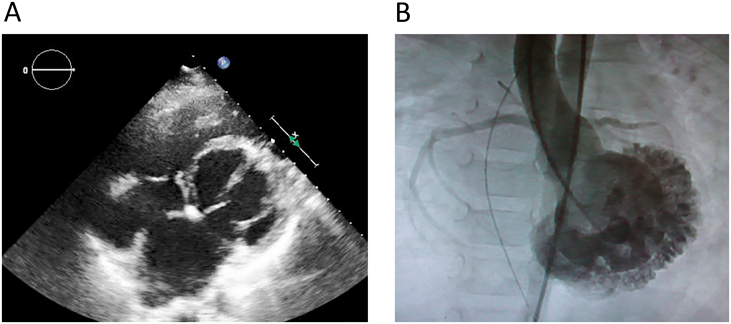
(A) The echocardiographic image. (B) The angiographic image. Note the both images showing lack of left ventricular apex and abnormal trabeculation of left ventricle.
約半数の小児RCM症例では心筋構造タンパクやイオンチャネル,細胞接着装置や細胞外基質をコードする遺伝子において,綿密な全エクソン解析を行っても有意な変異バリアントを同定できない.また変異が同定できた症例もできなかった症例も,心エコー所見を含む臨床像については明らかな差がなく,RCM表現型としては変異の有無で見分けがつかない.もちろん全ゲノムシークエンスを行うことによってエクソン内ではないゲノム異常を同定できる可能性はありうるが,そもそも心筋細胞にだけ着目していては心筋症の病態全てを解明できない可能性があると我々は考えている.そこで我々は,心筋細胞だけではなく心筋線維芽細胞にも着目してRCM病態解明の研究を行っている.心臓を構成する細胞は心筋細胞の他,約60%は心筋線維芽細胞が占めるとされ,近年,心筋線維芽細胞は,心筋の機能維持や線維化などに重要な役割を果たすことが明らかとなっている.しかし,特発性心筋症の病態における心筋線維芽細胞の役割は明らかになっていない.我々は小児RCM患者から単離した心筋線維芽細胞を用いて,その細胞生理学的機能や遺伝子発現プロファイルについて検討した.まず,細胞の生理学的基本機能である増殖能や遊走能,接着能,アポトーシスについては,健常心筋線維芽細胞とRCM患者心筋線維芽細胞とでは有意な差を認めなかった.しかし興味深いことに,次世代シークエンサーを用いた網羅的遺伝子発現解析(RNA-seq)では,RCM心筋線維芽細胞は健常心筋線維芽細胞とは全く異なった遺伝子発現プロファイルを呈しており,それはTNNI3変異の有無とは関連しなかった.すなわち心筋細胞で発現するトロポニンの異常があろうがなかろうが,RCM心筋線維芽細胞は正常とは異なるということである.次に,これらの心筋線維芽細胞を健常心筋細胞と共培養し,心筋細胞の拍動ベクトルをSONY SI8000モーションアナライザーを用いて解析した.驚くべきことに,RCM心筋線維芽細胞と共培養された健常心筋細胞は,その拡張能が増悪することが判明した.RCM心筋線維芽細胞では,心筋細胞の機能維持に重要とされるいくつかのケモカインやサイトカイン,細胞外基質の発現が亢進あるいは低下しており,心筋線維芽細胞自身が心筋細胞の拡張能増悪に寄与していることが明らかとなった16).今後,心筋線維芽細胞から分泌され心筋細胞の拡張能を増悪させる特定の因子を同定し,また,single cell RNA-seq解析によって,病態形成に深くかかわる心筋線維芽細胞のsub-population解析を行っていく予定である.また,我々はTNNI3変異を有する小児RCM患者からすでにiPS細胞も樹立している.これら患者iPS細胞から分化誘導したRCM心筋細胞とRCM心筋線維芽細胞との双方向性相互作用を明らかにしていくことにより,より詳細にRCM病態メカニズムの解明を行いたい.今後,心筋症の新たな治療ターゲットには心筋細胞だけではなく,その支持細胞の機能や心筋細胞との相互作用を制御するということも視野に入ってくるかもしれない.
小児期発症RCMは非常に予後が悪く,また,エビデンスのある治療法も存在しない.病態の進行に伴い,体液管理,心拍出量の維持,他臓器不全(肝うっ血,肺高血圧など)のコンロトールが極めて困難となってくる.また,機械的補助循環の安定した管理も困難を極めるため,進行症例では適切なタイミングで心臓移植につなぐことが肝要である.今後RCMの病態形成に関わる基礎研究が進むことによって,単にRCMだけにとどまらず,心室拡張障害そのものを改善させる治療法の開発に繋がってくることが期待される.
本論文において,開示すべき利益相反(COI)はない.
1) Webber SA, Lipshultz SE, Sleeper LA, et al: Pediatric cardiomyopathy registry investigators: Outcomes of restrictive cardiomyopathy in childhood and the influence of phenotype: A report from the Pediatric Cardiomyopathy Registry. Circulation 2012; 126: 1237–1244
2) Anderson HN, Cetta F, Driscoll DJ, et al: Idiopathic restrictive cardiomyopathy in children and young adults. Am J Cardiol 2018; 121: 1266–1270
3) Hayashi T, Tsuda E, Kurosaki K, et al: Electrocardiographic and clinical characteristics of idiopathic restrictive cardiomyopathy in children. Circ J 2007; 71: 1534–1539
4) Muraji S, Sumitomo N, Imamura T, et al: Diagnostic value of P-waves in children with idiopathic restrictive cardiomyopathy. Heart Vessels 2021; 36: 1141–1150
5) Ryan TD, Madueme PC, Jefferies JL, et al: Utility of echocardiography in the assessment of left ventricular diastolic function and restrictive physiology in children and young adults with restrictive cardiomyopathy: A comparative echocardiography-catheterization study. Pediatr Cardiol 2017; 38: 381–389
6) Tariq M, Ware SM: Importance of genetic evaluation and testing in pediatric cardiomyopathy. World J Cardiol 2014; 6: 1156–1165
7) Mogensen J, Kubo T, Duque M, et al: Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Invest 2003; 111: 209–216
8) Kostareva A, Kiselev A, Gudkova A, et al: Genetic spectrum of idiopathic restrictive cardiomyopathy uncovered by next-generation sequencing. PLoS One 2016; 11: e0163362
9) Hayashi T, Tanimoto K, Hirayama Yamada K, et al: Genetic background of Japanese patients with pediatric hypertrophic and restrictive cardiomyopathy. J Hum Genet 2018; 63: 989–996
10) Araki K, Ueno T, Taira M, et al: Pediatric patient with restrictive cardiomyopathy on staged biventricular assist device support with Berlin Heart EXCOR underwent heart transplantation successfully: The first case in Japan. J Artif Organs 2021; 24: 269–272
11) Su JA, Menteer J: Outcomes of Berlin Heart EXCOR pediatric ventricular assist device support in patients with restrictive and hypertrophic cardiomyopathy. Pediatr Transplant 2017; 21: e13048
12) Wittekind SG, Ryan TD, Gao Z, et al: Contemporary outcomes of pediatric restrictive cardiomyopathy: A single-center experience. Pediatr Cardiol 2019; 40: 694–704
13) Fernandez Valls M, Srichai MB, Stillman AE, et al: Isolated left ventricular apical hypoplasia: A new congenital anomaly described with cardiac tomography. Heart 2004; 90: 552–555
14) Meng H, Li JR, Sun X: Left ventricular apical hypoplasia: A case series and review of the literature. Acta Cardiol 2013; 68: 339–342
15) Skidan VI, Kuznetsova T, Pavlyukova EN, et al: Isolated left ventricular apical hypoplasia with myocardial non-compaction: A case report. Eur Heart J Case Rep 2020; 4: 1–6
16) Tsuru H, Ishida H, Narita J, et al: Cardiac fibroblasts play pathogenic roles in idiopathic restrictive cardiomyopathy. Circ J 2021; 85: 677–686


This page was created on 2021-11-26T15:07:40.963+09:00
This page was last modified on 2021-12-17T10:47:46.000+09:00
このサイトは(株)国際文献社によって運用されています。