先天性QT延長症候群(LQTS)は心筋イオンチャネルをエンコードする遺伝子異常により,心電図上QT延長やtorsade de pointes(TdP)と呼ばれる多形性心室頻拍を生じ,心臓突然死の原因となる疾患である1).LQTSはこれまで17個の原因遺伝子が報告されているが,同定される遺伝子型のほとんどがLQT1~3である1).
Long QT syndrome type 8(LQT8)は,QT延長に加えて,合指,先天性心疾患,精神発達遅滞など多臓器に所見を認めるTimothy症候群が知られている2).Timothy症候群は,染色体12p13.3にあるL型カルシウム(Ca)チャネル蛋白をコードする遺伝子(CACNA1C)のバリアントによって起こる症候群で,Splawskiらは,CACNA1Cのミスセンス変異(p.Gly406Arg)で生じる症候群をTimothy症候群として初めて報告した3).その後合指を認めないLQT8(Timothy症候群2)が報告され4),さらに心外合併症を認めないLQT8も報告されるようになった5–11).
今回我々は心外合併症を認めないLQT8の親子例を経験し,本患者に対してプロプラノロール,メキシレチン,ベラパミルの薬剤負荷試験を施行しQT時間の変化を評価したので報告する.
症例
18歳,男性
既往歴
特記事項なし
家族歴
父が健診でQT延長を指摘.父方曾祖父の弟が21歳時マラソン後に突然死.
病歴
小学校1年生の学校検診でQT延長を指摘され前医を受診した(Fig. 1).9歳時に遺伝子検査を実施されたが,Sanger法によるLQT1~3の遺伝子変異は認めなかった.13歳時に転居を機に当科を紹介受診した.以後当科で1年ごとの定期検診を受けていた.動悸,失神の既往はない.
生活歴
大学1年生
身体所見
身長170.0 cm,体重51.4 kg,脈拍82回/分,血圧139/82 mmHg, SpO2 99%(室内気)
顔貌正常(薄い髪の毛なし,耳介低位なし,鞍鼻なし,薄い上口唇なし,丸い顔貌なし),呼吸音清,心音整,心雑音なし,腹部平坦,軟,合指なし
検査所見
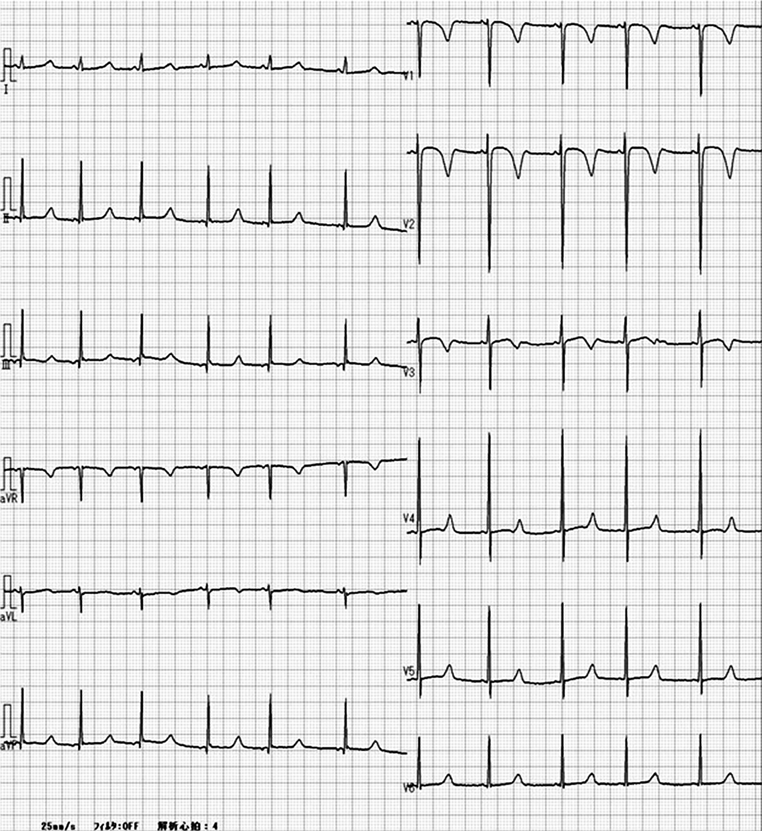
安静時心電図(Fig. 2A):心拍数58 bpm,洞調律,正軸,QTc(B: Bazett)436 msec, QTc(F: Fridericia)437 msec
経過
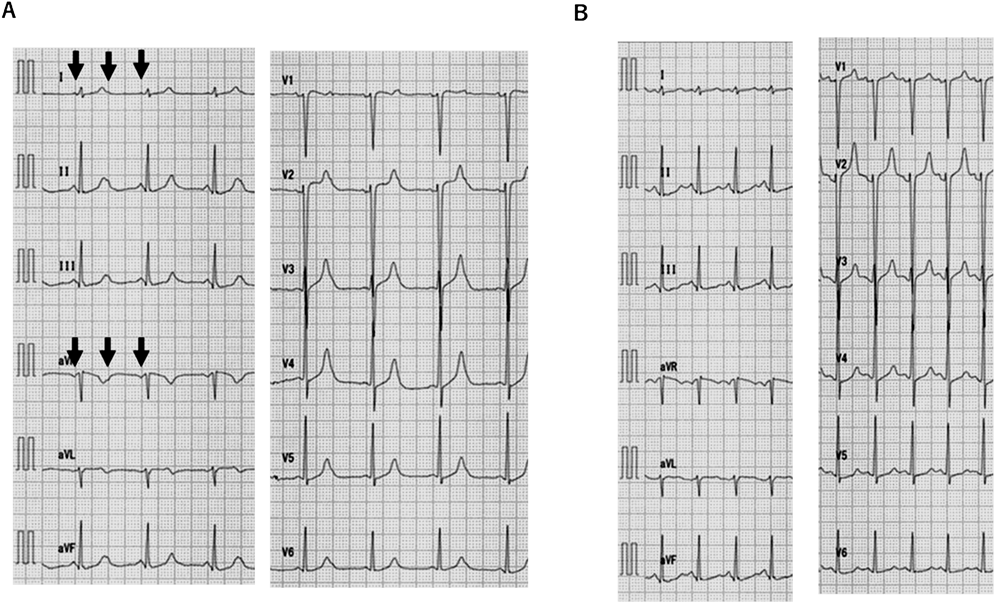
定期外来で運動負荷心電図検査(ダッシュ法)(protocol: manual,傾斜10%,速度10.5 km/h,1分間)を施行した.運動負荷中に心房レート150 bpmを越えた際に2 : 1房室ブロックを認め心室レートが150/分から75/分に突然低下したため検査を終了した.負荷直後の心電図(Fig. 3A)はP波がT波上にあり機能的2:1房室ブロックを来し(PP 386 msec), QT 380 msec, RR 820 msec, QTc(B)442 msec, QTc(F)427 msecとQT延長を認めず,負荷後35秒経過し心房レート130~140/分で1:1房室伝導に復帰した際の心電図(Fig. 3B)はQT 360 msec, RR 480 msec, QTc(B)520 msec, QTc(F)460 msecと延長を認めた.
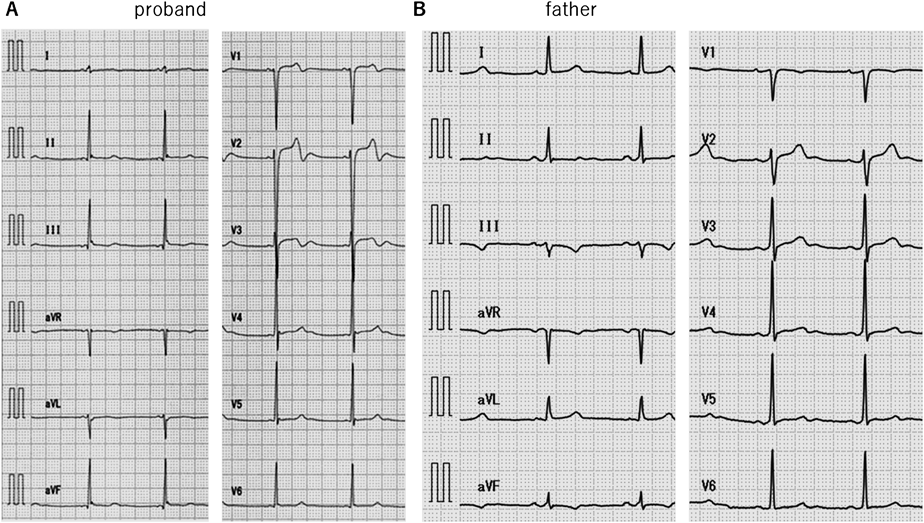
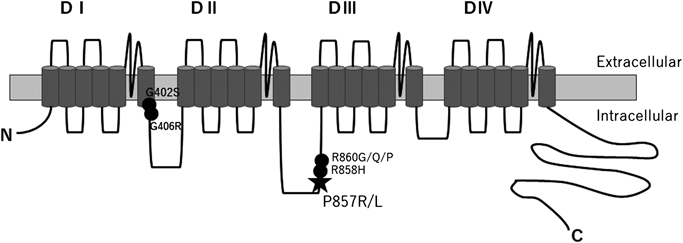
本患者と両親,弟に対して全エクソン遺伝子解析を施行した12)結果,本患者と父にLQT8の表現型として既報のCACNA1C遺伝子バリアント(c.2570C>G, p.Pro857Arg)が同定された.(Fig. 4)
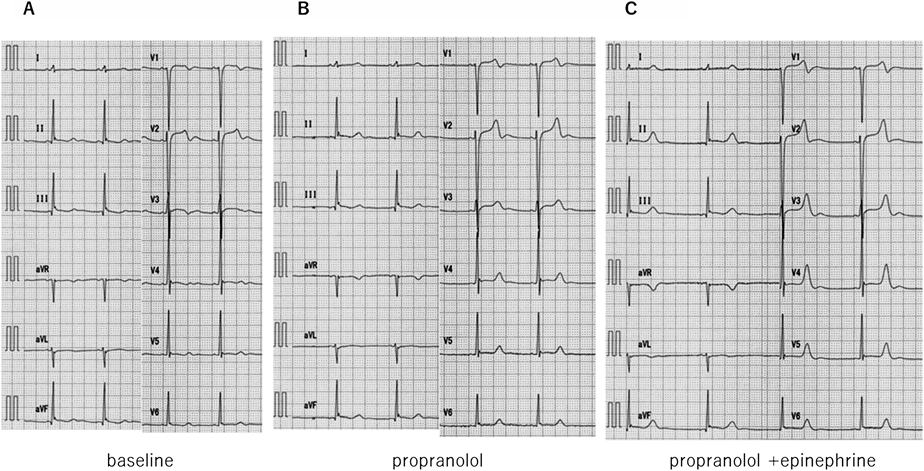
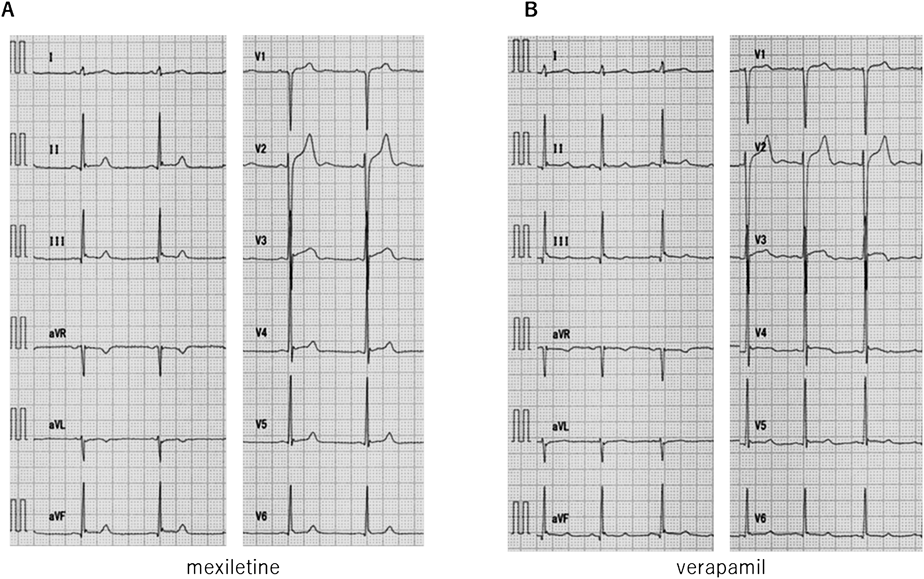
本患者に対してアドレナリン(エピネフリン)負荷試験,顔面浸水試験,プロプラノロール・メキシレチン・ベラパミルの薬剤負荷試験を施行しQT時間の変化を評価した.負荷試験の結果を表に示す(Table 1).
Table 1 Results of loading test | 負荷前 | 負荷後 |
|---|
| HR (bpm) | QT (msec) | QTc(B) (msec) | QTc(F) (msec) | HR (bpm) | QT (msec) | QTc(B) (msec) | QTc(F) (msec) |
|---|
| エピネフリン | 72 | 440 | 483 | 468 | 81 | 440 | 511 | 486 |
| 顔面浸水 | 58 | 475 | 468 | 470 | 52 | 490 | 454 | 466 |
| プロプラノロール | 71 | 470 | 513 | 498 | 60 | 470 | 470 | 470 |
| メキシレチン | 61 | 446 | 451 | 449 | 62 | 370 | 376 | 374 |
| ベラパミル | 62 | 420 | 429 | 426 | 81 | 360 | 418 | 398 |
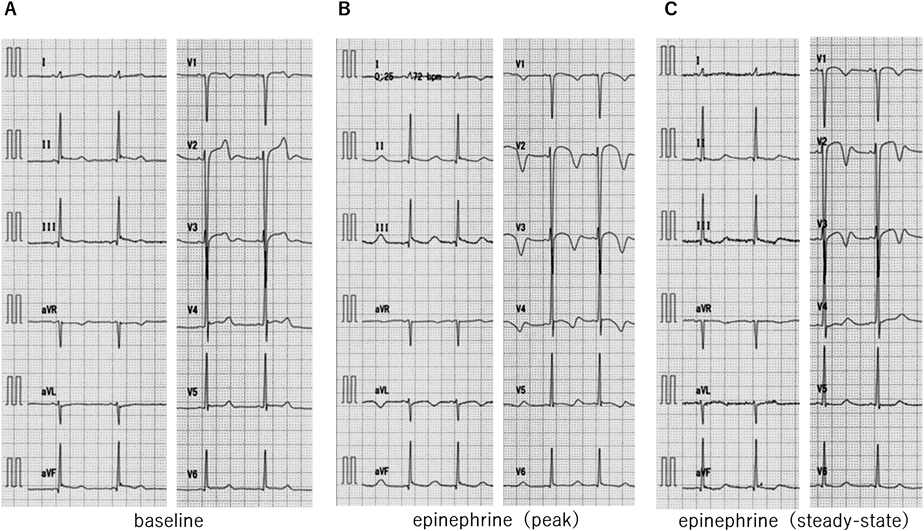
エピネフリン負荷試験は,清水らの方法13)に則り12誘導心電図をとりながら0.1 µg/kgをボーラス投与し,その後0.1 µg/kg/minの持続投与を5分間行った.エピネフリン投与を中止してさらに10分間心電図を記録した.投与開始後1~2分で二相性T波の顕在化を認めた.定常状態での心電図を負荷後の心電図とし,QTc(B)511 msec, QTc(F)486 msecと著しいQTc時間の延長を認めた(Fig. 5).顔面浸水試験は,心電図を記録しながら10°Cの冷水に顔面浸水を行った.息こらえの続く限り顔面浸水を持続し,浸水時間は37秒間であった.最大徐脈時を負荷後の心電図とし,徐脈時にQT延長の増悪は認めなかった.
プロプラノロール負荷試験は,12誘導心電図を記録しながらプロプラノロール0.1 mg/kgを10分間で投与した.投与終了時を負荷後の心電図とした.最大徐脈時はHR 32 bpm, QT 496 msec, QTc(B)364 msec, QTc(F)404 msecとQT延長は認めなかった.プロプラノロール投与終了5分後から,エピネフリン負荷試験を引き続いて行った(清水法)が,エピネフリン投与終了1分後にHR 47 bpm, QT 510 msec, QTc(B)447 msec, QTc(F)467 msecとβ遮断薬投与下ではエピネフリン負荷によるQT延長は抑制された(Fig. 6).
メキシレチン負荷試験は,12誘導心電図を記録しながらメキシレチン2 mg/kgを10分間で投与した.投与終了時にQTc(B)376 msec, QTc(F)374 msecとQTc時間の短縮を認めた(Fig. 7A).一方,ベラパミル負荷試験は,12誘導心電図を記録しながらベラパミル0.1 mg/kgを10分間で投与した.投与終了時はQTc(B)418 msec, QTc(F)398 msecであり,QTc時間は投与前と著変なかった(Fig. 7B).
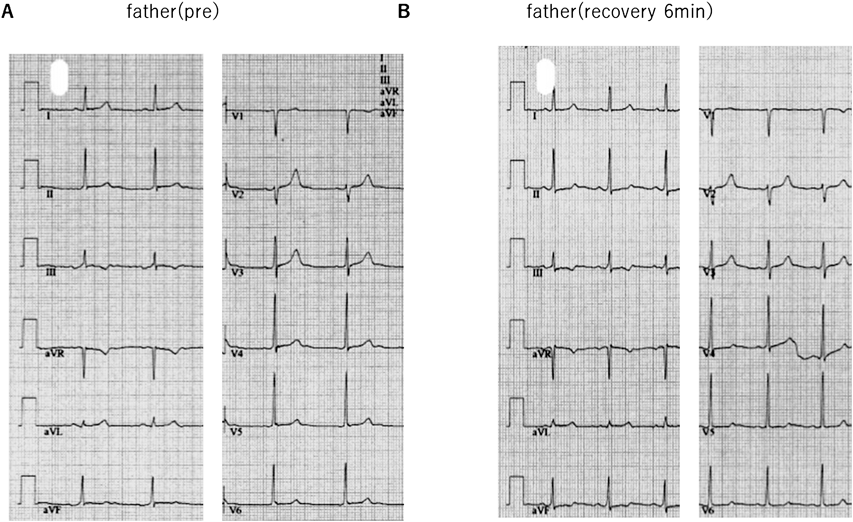
本患者の父親の安静時心電図(Fig. 2B)は心拍数60 bpm,洞調律,正軸,III陰性T波,QTc(B)429 msec, QTc(F)426 msecであった.父親に運動負荷心電図検査(Protocol: Bruce, StageIII9分,peak HR 151 bpm)を施行した(Fig. 8).負荷後6分で最大QTc(B)447 msec, QTc(F)431 msecと明らかなQT延長は認めず,機能的2 : 1房室ブロックも認めなかった.
本症例は学校心臓検診の心電図検査で初めてQT延長を指摘された.小学生当時は,QTc(B)≧500 msecで突然死の家族歴があり,Schwartzのリスクスコア14)は3.5点で診断確実であった.そこで,9歳時に遺伝子検査を施行したがLQT1~3の遺伝子変異は認めなかった.Schwartzのリスクスコアが3.5点のため,LQT1~3の変異は認めなかったものの,それ以外の先天性LQTSの可能性を考え,運動負荷心電図での1~2年ごとの定期検査を継続した.当院に通院するようになった13歳以降は,安静時心電図のQTc(F)は450~480 msec程度であり,当初よりQTは短縮傾向であった.しかし18歳時の運動負荷心電図でLQT8に特徴的2)な機能的2 : 1房室ブロックを認めたことを契機に遺伝子検査を再確認し,LQT8の診断に至った.
LQT8は,L型Caチャネル蛋白をコードするCACNA1C遺伝子異常によって生じる.CACNA1C遺伝子異常はL型Caチャネルの機能獲得型障害を起こし,心筋活動電流のうちCa電流を増強し,活動電位持続時間を延長させる15).LQT8が機能的2 : 1房室ブロックを生じるのは,房室結節よりも心室筋のほうが再分極時間が延長することが原因だと考えられている16).LQT8はSplawskiらにより,QT延長,合指,先天性心疾患,免疫不全,間欠的な低血糖,精神発達遅滞など多臓器に所見を認めるTimothy症候群として報告され,2004年にp.Gly406Arg(c.1216G>A),2005年にp.Gly402Ser(c.1204G>A)の遺伝子バリアントが報告されている3, 4).Splawskiらが2005年に報告した2症例はいずれも合指を認めず,合指を認めない症例はTimothy症候群2とされた.さらに心外合併症を認めないLQT8がBoczekらによって2013年に報告され,以降多数報告されている(Table 2).
Table 2 CACNA1C variants without any other extra-cardiac manifestations | 症例 | QTc (msec) | DNA | protein | LQT以外の心合併症 | 家族歴 |
|---|
| 本症例 | 18歳 男 | 437 | c.2570C>G | p.Pro857Arg | なし | 突然死+ 同変異+ |
| Colson et al, 2019 | 22歳 女 | 640 | c.1220A>C | p.Glu407Ala | 1度AVB | 突然死− 同変異− |
| Landstorm et al, 2016 | 6歳 女 | 530 | c.2284C>T | p.Leu762Phe | なし | 突然死+ 同変異+ |
| Boczek et al, 2015 | 33歳 女 | 500 | c.1552C>T | p.Arg518Cys | VSD,1度AVB,周産期心筋症,HCM洞結節機能不全 | 突然死+ 同変異+ |
| 10歳 男 | 480 | 同上 | 同上 | 突然死+ 同変異+ |
| 25歳 女 | 500 | c.1553G>A | p.Arg518His | Cleft mitral valve, ASD, PAC | 突然死+ 同変異+ |
| Hiippala et al, 2015 | 13歳 女 | 460 | c.1204G>A | p.Gly402Ser | なし | なし |
| Wemhoner et al, 2015 | 12歳 女 | 450 | c.82G>A | p.Ala28Thr | なし | 同変異+ |
| 30歳 男 | 498 | c.2578C>G | p.Arg860Gly | なし | なし |
| 15歳 女 | 452 | c.3496 A>G | p.Ile1166Val | なし | 同変異+ |
| 14歳 女 | 473 | c.4425C>G | p.Ile1475Met | なし | 同変異+ |
| 26歳 女 | 480 | c.4486G>A | p.Glu1496Lys | なし | なし |
| Fukuyama et al, 2014 | 8歳 女 | 480 | c.1141C>T | p.Pro381Ser | なし | 突然死− 同変異+ |
| 12歳 女 | 464 | c.1368G>A | p.Met456Ile | なし | 突然死− |
| 12歳 女 | 597 | c.1745C>A | p.Ala582Asp | 徐脈 | 突然死− 同変異+ |
| 54歳 女 | 435 | c.2339G>A | p.Arg858His | なし | 突然死+ 同変異+ |
| 7歳 男 | 476 | 同上 | 同上 | なし | 突然死+ |
| 15歳 男 | 420 | 同上 | 同上 | 徐脈 | 突然死− |
| 58歳 女 | 449 | c.5347G>T | p.Gly1783Cys | なし | 突然死+ |
| Boczek et al, 2013 | 27歳 女 | 498 | c.2570C>G | p.Pro857Arg | なし | 突然死+ 同変異+ |
| 15歳 女 | 475 | c.2500A>G | p.Lys834Glu | なし | なし |
| 15歳 男 | 514 | c.2570C>T | p.Pro857Leu | なし | 突然死+ 同変異+ |
| ASD, atrial septal defect; HCM, hypertrophic cardiomyopathy; PAC, premature atrial contraction; VSD, ventricular septal defect |
本症例に認めたバリアントp.Pro857Arg(c.2570C>G)はBoczekらにより2013年に初めて報告され無症状であった5).我が国では最近大野らが25症例のLQT8を報告し,9例が失神または心肺蘇生後であるが16例は無症状であり,Pro857Argバリアントは1例認められ無症状であった17).
今回,本症例の安静時QTcは450~480 msec前後であった.運動負荷時やエピネフリン負荷時にQT延長の増悪を認め,遺伝性不整脈の診療に関するガイドライン14)で高リスクとされる500 msec以上にも達していた.また,Giudicessiらは先天性LQTSの遺伝子型と表現型に基づくリスク層別化をレビューしており,40歳までに心事故が発生するリスクを,Extremely High Risk(≧80%),High Risk(≧50%),Intermediate Risk(30~49%),Low Risk(<30%)に層別化している18).このレビューでTimothy症候群はExtremely High Risk群に分類されているが,Timothy症候群型LQT8はGly406ArgとGly402Serに限られており,本症例はCACNA1C遺伝子にバリアントがあるもののTimothy症候群ではなく,安静時QTc<500 msecのためLow Risk群に分類される.また家系解析でも父親が同バリアントを有しているものの,安静・負荷心電図ともに明らかなQT延長はないことからも,本バリアント単独での病的意義には疑問が残る.また大野らの報告でもPro857Argは心電図のT波形態から比較的低リスク型の可能性が示唆される17).
一方でMellorらは非症候性のLQT8患者23人から9種類のCACNA1C遺伝子の病的バリアントもしくは病的と思われるバリアントを同定し19),この9種類のバリアントのうち6種類(p.Pro857Arg, p.Pro857Leu, p.Arg858His, p.Arg860Gly, p.Arg860Gln, p.Arg860Pro)がL型Caチャネルの857~860の4つのアミノ酸配列,すなわちCaチャネルのαサブユニットのII–III cytosolic loop内の小領域に該当した(Fig. 9).II–III cytosolic loop内にバリアントがあった患者群はそれ以外の部分にバリアントがあった患者群よりQTc時間が短い(477±31msec vs. 515±37 msec,p=0.03)にもかかわらず,不整脈イベントの発生は同程度であった(53% vs. 48%,p=0.41).また,突然死の家族歴はII–III cytosolic loop内にバリアントがあった患者群のみに認められた(60% vs. 0%,p=0.03).以上から本症例もPro857Argであり,安静時QTcが500 msec未満であっても必ずしも不整脈イベントや突然死のリスクが低いとはいえないと考えられる.
Timothy症候群と呼ばれるLQT8はGly406ArgあるいはGly402Serが代表的でTdPや心室細動などを来たす予後不良な疾患とされている.治療法としてICDの有効性が期待されている15)が,新生児・乳幼児などの体格の小さい患者や,本症例のように致死性の不整脈イベントのない患者に対しては,ICDを積極的には選択しにくい.LQT8に対する抗不整脈薬の予防内服に関してはβ遮断薬,メキシレチン,ベラパミルなどの報告が散見されるが,明確なエビデンス,治療指針はない.
LQT8の突然死予防に第一選択として用いられることが多い薬剤はβ遮断薬である20, 21).交感神経刺激の抑制による致死性不整脈の予防や,徐拍化による機能的2:1房室ブロックの改善効果が期待されるが,β遮断薬単剤での有効性の報告は少数である22, 23).LQT8に対してのβ遮断薬は,単剤ではなくメキシレチンを併用することでQT時間が短縮して有効だったとの報告がある24–27).メキシレチンは単剤での有効性の報告もある16).
LQT8はL型Caチャネルの機能獲得型変異が原因であることから,ベラパミルなどCa拮抗薬の効果が期待されてきた14).Jacobらは,心室細動と心停止の既往でICD(埋め込み型除細動器)植込み後の21歳男性(p.Gly402Ser)にベラパミルを開始しICD作動を減少させることができたと報告している28).しかし,LQT8に対するCa拮抗薬の効果は否定的な報告も散見される16, 26, 29).本症例へのベラパミル投与でQT延長の明らかな改善は認めなかったことからも,LQT8に対するベラパミルなどのCa拮抗薬の投薬は慎重にすべきと考えられる.
本症例では,プロプラノロール投与下のエピネフリン負荷で,エピネフリン投与終了1分後で最大QTc(B)447 msec, QTc(F)467 msecとQT延長はある程度抑制された.またメキシレチン投与ではQTc(B)376 msec, QTc(F)374 msecとQTc時間の短縮を認めた.本症例はこれまで無症状だが,protocolがmanual(1分間で急速に心拍数を上げるダッシュ法)の運動負荷心電図で機能的房室ブロックを認めた.運動負荷心電図は1~2年に1回の頻度で施行しており,LQT8の診断後も再度ダッシュ法とBruce法を施行したが,機能的房室ブロックは認めず再現性は低かった.しかしながら突然死の家族歴があることを踏まえ,激しい運動や競技,体調不良時の運動は制限する方針で管理されている.今後予防内服を検討する場合はプロプラノロールとメキシレチンの併用,もしくはメキシレチン単剤の有効性が期待される.