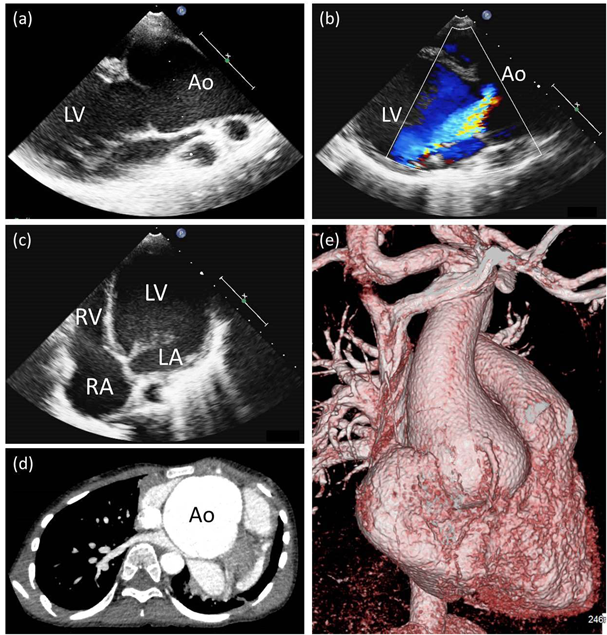
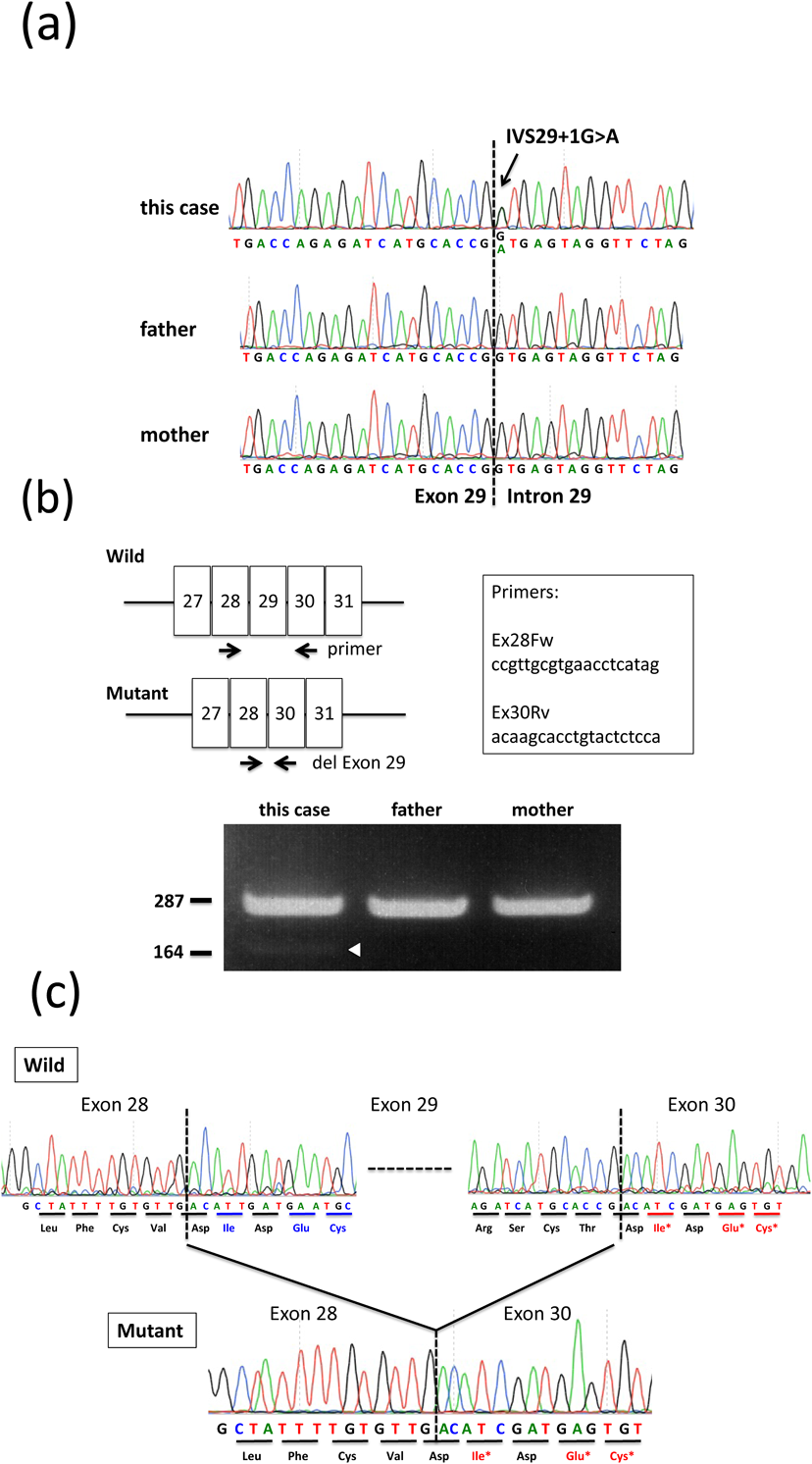
FBN1遺伝子第29番エクソンのスプライシング異常による早期発症型Marfan症候群の一例Early-onset Marfan Syndrome Caused by a Splicing Mutation of FBN1 Exon 29: A Case Report
1 東京大学医学部附属病院小児科Department of Pediatrics, The University of Tokyo ◇ Tokyo, Japan
2 東京大学医学部附属病院循環器内科Department of Cardiovascular Medicine, The University of Tokyo ◇ Tokyo, Japan
3 東京大学医学部附属病院整形外科Department of Orthopedic Surgery, The University of Tokyo ◇ Tokyo, Japan
受付日:2017年11月29日Received: November 29, 2017
受理日:2018年2月14日Accepted: February 14, 2018
発行日:2018年3月1日Published: March 1, 2018