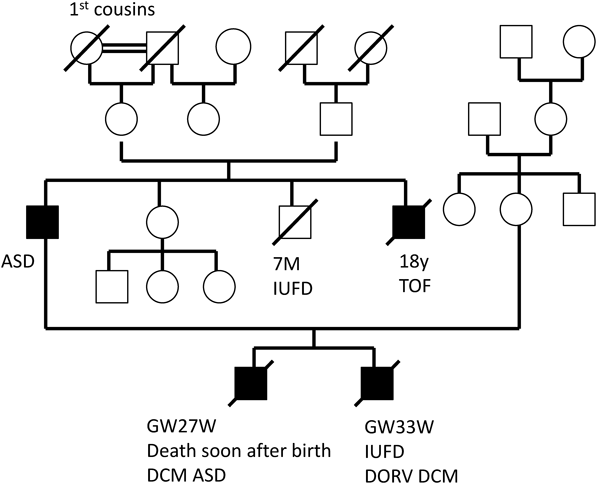
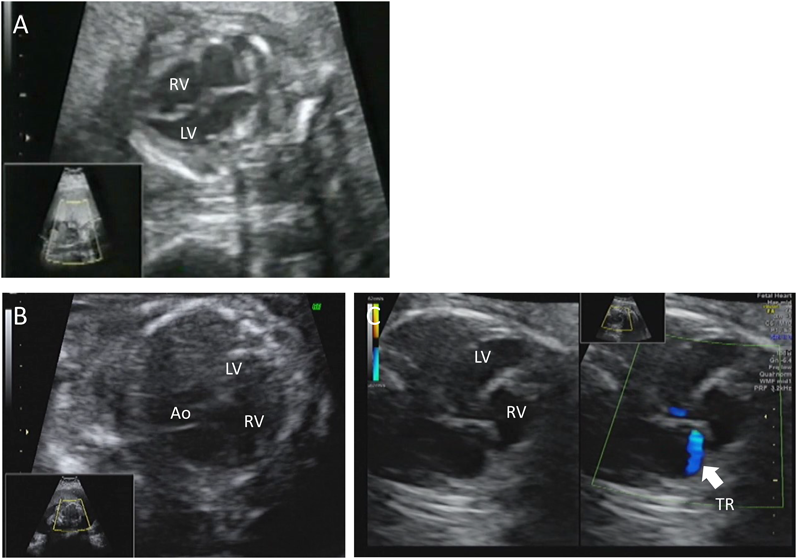
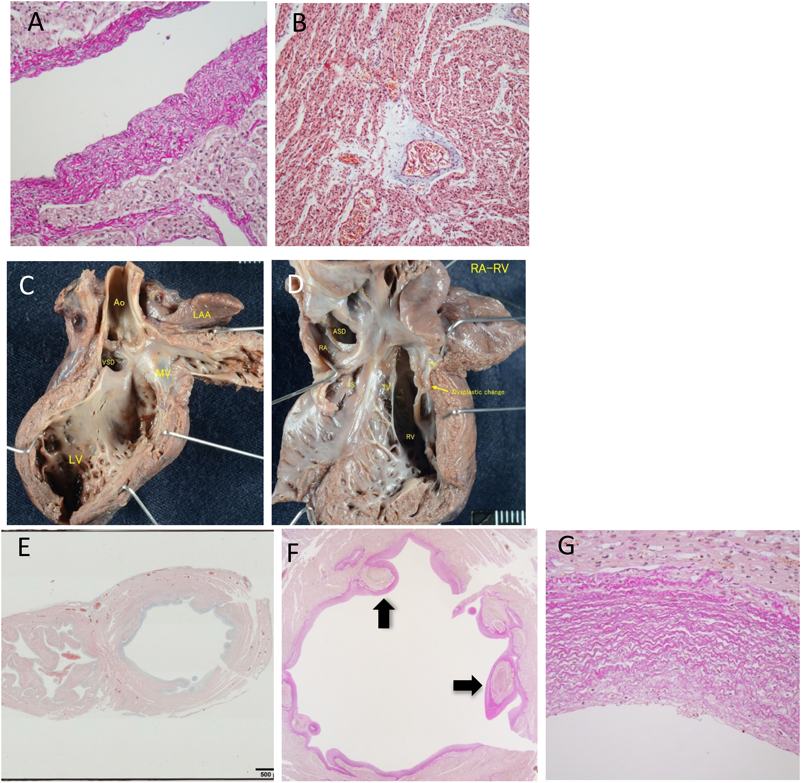
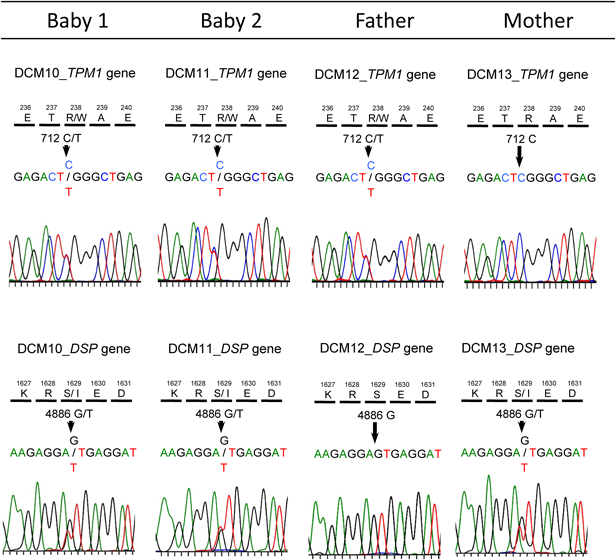
胎児期より拡張型心筋症と診断され周産期死亡した兄弟例Fetal Sibling Case of Familial Dilated Cardiomyopathy
1 兵庫県立こども病院循環器科Department of Cardiology, Hyogo Children’s Hospital ◇ Hyogo, Japan
2 日赤和歌山医療センター小児科Department of Pediatrics, Japanese Red Cross Wakayama Medical Center ◇ Wakayama, Japan
3 加古川医療センター小児科Department of Pediatrics, Kakogawa Central City Hospital ◇ Hyogo, Japan
4 富山大学医学部小児科Department of Pediatrics, Toyama University Hospital ◇ Toyama, Japan
5 富山大学医学部法医学Department of Legal Medicine, Graduate School of Medicine University of Toyama Hospital ◇ Toyama, Japan
受付日:2017年10月19日Received: October 19, 2017
受理日:2017年12月21日Accepted: December 21, 2017
発行日:2018年1月1日Published: January 1, 2018