生来健康であった9歳男児.学校心臓病検診で心電図異常を指摘された.学校検診の心電図では6歳時には異常を認めなかったが,9歳3か月での検査ではI誘導でQSパターン,心室性期外収縮(PVC)を認めた(Fig. 1).成長発達で異常を認めず,突然死や心筋症等の家族歴はなかった.学校心臓病検診受診の際には動悸,胸痛,易疲労感などの自覚症状は認められなかったが,9歳5か月頃には倦怠感と動悸を自覚するようになっていた.他院を受診時には心胸郭比67%と心拡大を認め,BNP1474 pg/mLと高値であった.心不全治療および不整脈治療として利尿薬,PDEIII阻害薬,β遮断薬,アミオダロンを開始されたうえで,PVCに対するカテーテル焼灼術目的に当院に転院となった.
身体所見
身長142.0 cm(+1.0 SD),体重32 kg(−0.1 SD).顔色不良で,眼瞼結膜は貧血様.胸部聴診では異常を認めず,肝臓は1 cm程度触知した.浮腫は認めなかった.
検査所見
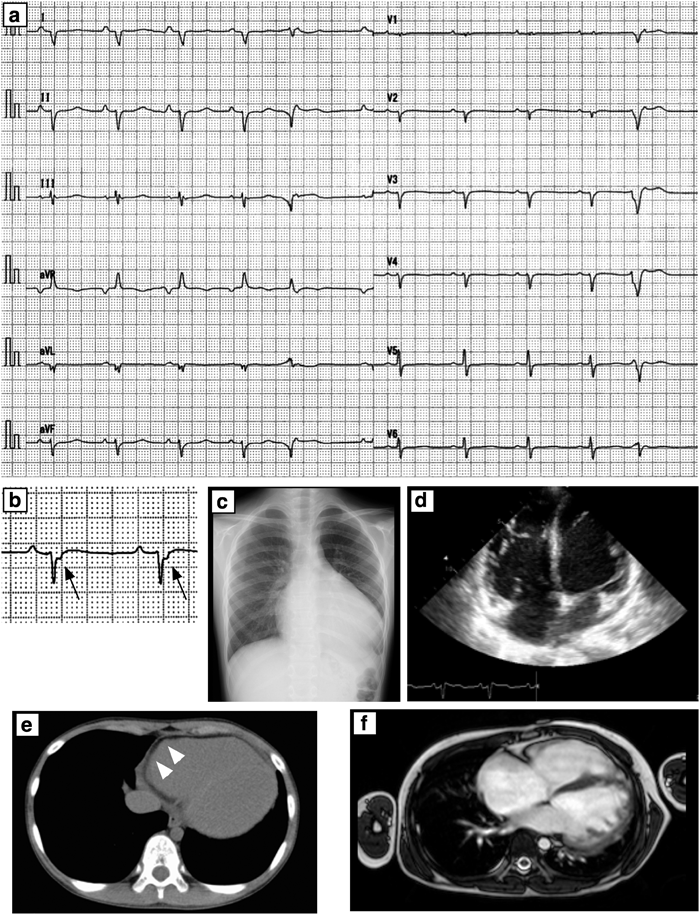
12誘導心電図ではQRS軸が不定軸で低電位,V1–3誘導においてはイプシロン波を認めた(Fig. 2a, b).血液検査ではBNP 895.1 pg/mLと高値を認めたが,その他の血液生化学所見には異常を認めなかった.Holter心電図では右室起源の多源性心室期外収縮と13連発のnon-sustained VTを認めた.遅延電位ではfQRS 173 ms(>114 ms),RMS40 3.0 µV(<20 µV),LAS 88.5 ms(>38 ms)と異常を認めた.胸部単純X線では心拡大を認めた(CTR 67%)(Fig. 2c)心エコーでは両心室の著明な拡大,左室収縮能の著明な低下(LVFS 0.04)と軽度の僧帽弁逆流を認めた(Fig. 2.d).心肺運動負荷試験では最大酸素摂取量15.4 mL/kg/min(予測値の38%),無酸素性作業閾値10.3 mL/kg/min(予測値の26%)と機能低下を認めた.マルチスライスCTでは右室壁の脂肪変性を認めた(Fig. 2e).心臓MRIでは両心室で内腔の拡大と駆出率の低下(右室駆出率23%,右室拡張末期容積237 mL,左室駆出率17%,左室拡張末期容積179 mL)を認めた(Fig. 2f).
心臓カテーテルで電気生理学的焼灼及び心筋生検を予定したが,右室流出路でのカテーテル操作で心室頻拍が頻発し血行動態が悪化したため血行動態的評価のみで検査を終了した(Table 1).以上の結果より,改訂版Task Force Criteria6)よりARVCと診断した.
Table 1 Cardiac catheterization data | Pressure (mmHg) | O2 saturation (%) |
|---|
| Systolic | Diastolic / EDP | Mean |
|---|
| Right Atrium | — | — | 13 | 59 |
| Right Ventricle | 36 | EDP17 | — | — |
| Pulmonary Artery | 34 | 18 | 25 | 59 |
| PA Wedge | — | — | 13 | — |
| Left Ventricle | 77 | EDP15 | — | — |
| Cardiac Output | 2.4 | (L/min) |
| Cardiac Index | 2.3 | (L/min/m2) |
治療
心不全治療として利尿薬,β遮断薬,ACE阻害薬,アミオダロンの内服を他院で開始されていた.当院転院後,カテーテル後の心不全に対してミルリノン,ドブタミンによる治療を開始した.CRT-D(cardiac resynchronization therapy-defibrillator)も考慮したが,narrow QRSでありCRTの有効性が期待されないことと,カテーテル時に血行動態が悪化したこと,アミオダロンでPVCのコントロールがついたことから積極的適応はないと判断した.その後β遮断薬の増量のうえでもBNPは900~1,300 pg/mL程度で推移しており,カテコラミンの減量を試みるも困難な状況が続いたため心臓移植の適応と考えた.10歳0か月で院内の適応検討を経て,日本循環器学会心臓移植委員会で適応の判定を受けた.10歳9か月で渡航し1か月の待機で心臓移植を受けた.
摘出心において,肉眼的に両心室は著明に拡大し,右室壁は脂肪織に置換され高度の菲薄化を認めた.組織学的には,右室における線維脂肪変性を認め,島状に散在する残存心筋には肥大が認められた.左室では心筋細胞の肥大と斑状の線維化が目立ち,軽度に脂肪織を認めた(Fig. 3).炎症細胞浸潤は認められなかった.
遺伝的背景の検討
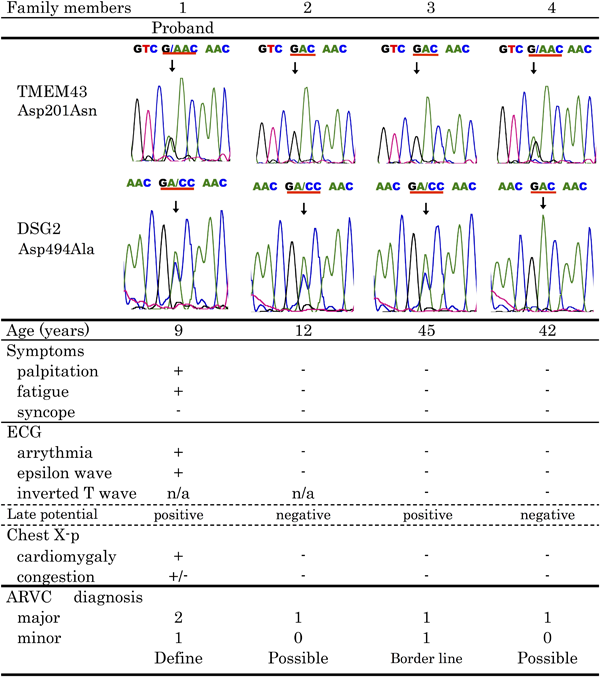
ARVC関連遺伝子変異の検索としてplakophilin 2(PKP2), desmoglein2(DSG2), desmoplakin(DSP), desmocollin 2(DSC2), transmembrane 43(TMEM43)において既報の変異を有するエクソンに関してダイレクトシークエンス法により変異の有無を検討した.患児ではDSG2とTMEM43の点突然変異をヘテロ接合で認めた(DSG2(c.1481 A>C),TMEM43(c.601G>A))(Table 2).
Table 2 Detailed descriptions of the genetic mutations detected in the patient| Gene | Protein | dbSNP | Frequency † | SIFT | PolyPhen-2 | PANTHER |
|---|
| subPSEC | Pdeleterious |
|---|
| DSG-2 c.1481 A>C | p.Asp494Ala | rs19329842 8 | 0.04% | damagin g | probably damaging | −2.23 | 0.317 |
| TMEM43 c.601G>A | p.Asp201As n | rs13818227 6 | 0.06% | tolerated | benign | −1.01 | 0.12 |
| dbSNP; data base of single nucleotide polymorphism on the National center for Biotechnology Information. †: Frequency in 1000 Genome phase 1 population. [Web resorce; NCBI dbSNP, SIFT, PolyPhen-2, Panther] |
左室病変が顕著であったため拡張型心筋症(dilated cardiomyopathy; DCM)に関連した遺伝子(cardiac Troponin T(TNNT2), beta-Myosin Heavy chain(MYH7), cardiac Myosin-Binding Protein C(MYBPC3), cardiac Troponin I(TNNI3), alpha-Tropomyosin(TPM1), cardiac alpha-Actin(ACTC), Myosin light chain 2(MYL2), Myosin light chain 3(MYL3), alpha-Tropomyosin(TPM1))に関しても検討を行ったがいずれの遺伝子においても病因となる変異は認められなかった.
家族の遺伝子検査も施行したところ,家族内でも患児と同じ遺伝子変異を認めた(Fig. 4).しかし,家族内でDSG2とTMEM43両方の変異を有するのは患児のみであった.また,家族の心電図,胸部X線,心エコー,遅延電位の評価を行ったところ,患児の父のみ心電図で心室性期外収縮を認め,遅延電位でも異常を認めた.Holter心電図では2カ所の起原を持つ心室性期外収縮を認めた.心臓MRIでは右心室のEFが軽度低下していた(RVEF41%).心臓カテーテル検査では冠動脈に異常を認めず,LVEF48%,RVEF39%であった.LVはseg4領域で収縮の低下を認め,RVはdiffuse hypokinesisであった.心筋生検では脂肪線維変性はみられなかったが心筋肥大,核の腫大・変性,間質の線維化を認めた.以上から患児の父はARVC borderlineと診断し,以後外来経過観察を行っている.
小児期発症のARVCは稀な病態であると考えられるが,本症例は学校心臓病検診を契機に9歳時に診断に至り,早期の治療介入が可能であった.本症例の特徴としては若年発症のARVCであることと,重篤な左心不全症状を発症早期から呈したことが挙げられる.
ARVCの原因としてデスモゾーム関連タンパク質(PKP2, DSG2, DSP, DSC2, junctional plakogrobin; JUP)や非デスモゾーム関連タンパク質(desmin; DES, TMEM43, titin; TTN, Lamin A; LMNA, phospholanban; PLN, ryanodine receptor2; RYR2, transforming growth factor beta1; TGFB1)の変異が報告されており,各遺伝子変異が発症年齢に与える影響に関しては種々の報告がある1, 3, 7).しかし,これらの報告の中でも20歳代での発症が論じられているにとどまる.
また,ARVCにおける左心系への病理学的変化の進展は発症からの経過年数に応じその程度・頻度が増加するとされており,若年者においては左心室での病理学的異常所見は少なく8),遺伝子としてはDSG2, DSC, PLN, TMEM43の異常がARVCにおける左心系への進行に寄与しているとされている9, 10).本症例ではDSG2とTMEM43に変異を認めたが,いずれもNational Center of Biotechnology Informations(NCBI)のデータベースにはsingle nucleotide polymorphism(SNP)として登録されているものであった(Table 2).しかし,Table 2に示すようにいずれのSNPも健常者における頻度は非常に低いものであり,ARVCは遺伝的心筋症でありながら発症年齢が高いことから健康成人を対象としたサンプル群から作成されたSNPデータベースには未発症例が混在している可能性はある.したがってSNPであっても病因変異である可能性は否定できない.
DSG2(c.1481 A>C)に関してはpolyphenやSIFT,PANTHERによる予測ではタンパク質機能異常をきたす可能性が高い変異であった.日本人のARVCに関する検討では本家系にみられたDSG2(c.1481 A>C)と同様の変異が報告されている1)ことから,DSG2(c.1481 A>C)に関してはARVC発症に寄与している可能性が高い.
ARVCによる若年突然死症例でのDSG2変異の症例報告はある11)が,本症例でみられた変異部位(c.1481 A>C)に関してはこれまでの報告では若年発症への寄与は示されていない.また,本家系において患児の父と姉が同様のDSG2の変異を有しているにもかかわらず発症に至っていないことからはDSG2(c.1481 A>C)自体が早期発症に関わっているかどうかの判断は困難である.他の保因者が未発症であることから,この変異が発症に至るかは浸透率やその他の遺伝的・環境的な関与があるものと推察される.この点に関して,本症例ではTMEM43にもアミノ酸変化を伴う変異を認めたことが発症に寄与した可能性を考慮する必要がある.
ARVC5の原因とされるTMEM43の変異(c.1073C>T: p.S358L)では変異型TMEM43の存在により,デスモゾーム構成タンパク質や細胞骨格構成タンパク質の発現に影響を与え,若年発症や左心系への病勢の進展に寄与するという報告がある.本家系におけるTMEM43の変異は単独保因者である母が発症に至っていない点やpolyphen,SIFT,PANTHERの結果からは少なくとも単独の変異で発症に至るものではないと考えられる.そのため,TMEM43(c.601G>A)の変異がデスモゾーム構成タンパク質の変異に影響を与えるか否かに関しては今後の症例の蓄積やタンパク質機能の解析を要する.DSG2(c.1481 A>C),TMEM43(c.601G>A)いずれにおいても今後機能解析により病態形成への寄与の有無を検討していく必要がある.また,遺伝子変異が認められた家族の心電図や心エコーも定期的に評価が重要と考える.
病因遺伝子の研究においては「変異のある遺伝子」=「疾患責任遺伝子」と断定できず,変異遺伝子のタンパク質機能解析を必要とする.これまでの研究は細胞株や実験動物によって行われてきた.これらの問題点は種差により,必ずしもヒトにおける表現型を担保しないという点にあったが,ヒト人工多能性幹細胞(iPS細胞)12)は患者由来の細胞系を用いた解析を可能とした.既にARVCにおいても患者由来iPS細胞の報告がなされ病因解明を期待されている13–15).
ARVCの責任遺伝子の多くがデスモゾーム構成タンパク質であり,いかにして脂肪変性が起きるか,なぜ右室に病変が優位であるのかその全容は明らかでない.しかし,心臓発生とARVCの病因が密接に関与しているとする興味深い仮説がある.
右心室とくに右室流出路を形成する心筋は二次心臓形成領域(secondary heart field; SHF)に由来し,Wnt/βカテニンシグナルがSHFの形成・維持に重要な役割を担っている16, 17).脂肪分化を誘導する転写因子PPARγは通常βカテニンにより抑制されているが,βカテニンと分子構造の類似したjunctional plakogrobinは核内でPPARγのプロモーター領域に結合し,βカテニンと対照的にPPARγ発現を促進する.デスモゾームの異常によりjunctional plakogrobinがデスモゾームに局在できなくなり,核内に移行することがPPARγの発現促進,脂肪変性へつながる.この影響が色濃く出るのがSHFに由来する右室心筋であるとする仮説18)である.しかし,これまでの報告ではDSPの発現抑制でこの現象が確認された19)のみであり,他のデスモゾーム構成タンパク質や非デスモゾーム構成タンパク質の変異によっても同様の現象が再現されるかは不明である.そのため,今後の研究では細胞接着や細胞骨格の評価に加えWnt/βカテニンシグナルの評価も行っていくことがタンパク質機能の解析およびARVCの病因究明に有用であると考えられる.