交感神経系およびレニン–アンギオテンシン–アルドステロン系
PAH患者では血中のカテコラミン値,レニン活性,アンギオテンシンIIやアルドステロン濃度が高値を示し,交感神経系の過緊張状態やレニン–アンギオテンシン–アルドステロン系(renin–angiotensin–aldosterone system, RAAS)の活性が報告されている1–3).欧州のPAHガイドラインではβ遮断薬とRAAS阻害薬に関して推奨はしていないが4),新たな治療薬も開発されており,この経路における薬剤は今後も試されていくと考えられる.
β遮断薬
PAH患者では心拍出量を維持するために,交感神経系の亢進によって心拍数を増加させる代償が働いている.このため,PAHに対してβ遮断薬は従来禁忌と考えられており,過去の報告では運動耐容能や肺循環動態への有効性は見いだせていない5).しかし,PAHの病態においてβ1受容体の活性化が関与しており,この受容体を遮断することで肺血管の平滑筋細胞の増殖抑制や炎症性サイトカイン分泌抑制による心筋線維化を抑制することが可能であり,さらにα1受容体の抑制によっても肺血管の攣縮を改善させることがわかってきた6).そのβ1, 2およびα1受容体を抑制する非選択的β受容体阻害薬のカルベジロールが,PAHモデルにおいて右室リモデリングや右心機能を改善させることが報告された7).そこで,PAH患者に対するカルベジロールの二重盲検ランダム化比較試験(randomized controlled trial, RCT)が行われたが,用量依存性(6.25~50 mg/日)に運動時の心拍数を減少させたものの体血圧は10 mmHg程度低下し,臨床的な有効性は得られなかった8).次に試されたβ遮断薬のビソプロロールでも忍容性はあるものの,カルベジロール同様に右心機能や予後において改善は認められなかった9, 10).また,PAH患者に対してメトプロロール,アテノロール,カルベジロールのいずれかを使用した過去の報告からメタ解析を行ったところ,明らかな臨床的増悪を来さないものの右心機能や予後は改善しないという結論であった11).
しかし,近年になり選択的β遮断薬であるネビボロールの有効性が示された.ネビボロールは選択的β1受容体遮断薬であると同時にβ3受容体の作動薬でもあり,血管内皮由来の一酸化窒素(nitric oxide, NO)産生の促進や,活性酸素種を除去する作用を併せ持つ12).この機序によってPAHにおいて血管内皮障害を改善し,肺血管のリモデリングや右心機能の改善を示すことが報告されており13),PAHの病態進展を抑制する治療薬の1つとして期待されている.
RAAS阻害薬
PAH患者ではレニン活性,アンギオテンシンIIやアルドステロンの濃度が高いことが知られており,RAAS阻害薬による治療効果が検討されている14, 15).しかし,アルドステロンの血中濃度は肺循環動態や重症度と相関はなく,RAAS阻害薬であるスピロノラクトン併用療法の検討でも臨床的改善は認められなかった16, 17).一方,ロサルタン治療で肺循環動態が改善したという報告があるが,少数例の検討であり十分なエビデンスがあるとは言えない18).
RAASに作用する物質で近年注目されているのがapelinである.Apelin(APJ endogenous ligand)は13のアミノ酸から成るペプチドで,APJ受容体(apelin receptor)に結合するアゴニストである.APJ受容体は肺,心臓,胎盤などの様々な組織に発現しており,血管拡張作用,心筋収縮力増強,体液調節,血管新生などに関与する19–21).APJ受容体はアンギオテンシンII1型受容体と高い相同性を示し,apelinが結合するとアンギオテンシン変換酵素2の発現促進とアンギオテンシンII1型受容体の抑制作用を示し,アンギオテンシンIIを阻害する.Apelin自体が強力な陽性変力作用を有し用量依存性に心筋細胞の収縮力を増加させるため,心不全治療薬になる可能性も示唆されている.他にもapelinはバソプレッシンの分泌抑制を示すなどRAASに対する強力な抑制作用を持つことで心保護に働く.また,apelinは血管内皮細胞に多く発現しており,NO産生を促進することで血管拡張作用を有する他,血管内皮細胞の増殖作用を持ち,血管新生に重要な役割があることもわかってきた.PAHにおける報告では,apelin欠損マウスで肺動脈圧が上昇することや肺動脈の内皮細胞でその発現が減弱していたことから,apelinがPAHの病態に関与していることが疑われる22).
APJ受容体に結合するもう一つのペプチドにelabelaがある.ElabelaはAPJ受容体と結合しapelinと似たような作用を示すが,心臓発生において重要な役割を持つことが知られており,apelinとは異なるAPJ受容体リガンドとして存在する23).Apelinとの関連性についてはまだ十分わかっていないが,PAHモデルへのelabela投与により右室圧の低下,右室や肺血管のリモデリングの改善がみられることから24),この経路に対する治療薬として期待されている.
肺動脈自律神経叢除神経療法(PADN)
肺動脈自律神経叢除神経療法(pulmonary artery denervation, PADN)とは,PAHにおける交感神経系の過緊張状態に対し,肺動脈に分布する自律神経叢へのカテーテル焼灼術を行う治療である.既に高血圧症例に対してカテーテルにより腎交感神経を焼却除神経する治療が行われていたが,これと同様の治療コンセプトでPAHモデルに対し主にPADNが施行された25).自律神経叢は主肺動脈の左側壁に存在しており,そこへ高周波電流(20~40 W)によるカテーテルアブレーションを行い,合計120秒以内の焼却時間で十分な効果が得られるようである.2013年に中国から報告されたPADN-1 study(pulmonary artery denervation for treatment of pulmonary artery hypertension)では,13例の特発性PAH(idiopathic pulmonary artery hypertension, IPAH)に対して行われた26).PADN治療3か月後の平均肺動脈圧が55 mmHgから36 mmHgへ低下し,6分間歩行距離やWHO(World Health Organization)肺高血圧症機能分類の改善を認め,重篤な周術期の合併症はなかった.その後もPADNによるPAHへの臨床試験は主に中国から報告されている27–29).治療は局所麻酔で行われており,術中の胸痛はほとんどの症例で認められる.想定される合併症として肺動脈穿孔,心タンポナーデ,出血,反回神経麻痺などが挙げられるが,そのリスクは高くないとされている.第II相臨床試験で66例のPAH患者に対する治療効果が検討されており27),94%の症例で平均肺動脈圧が10%以上低下した.この治療効果は1年後にも維持されていたが,経過観察中10例でPAH増悪または死亡があった.しかし,この研究の対象患者は治療前の平均肺動脈圧が40 mmHg前後であり,ほとんどがNYHA(New York Heart Association)機能分類でIIまたはIII度であったことから重症例ではリスクが高くなる可能性が考えられる.
経皮的心房中隔裂開術(balloon atrial septostomy, BAS)は重症PAHにおいて,右左短絡による心拍出量の増加を目的として行われてきた.成人PAHにおいて最大限の内科的治療に不応の場合,肺移植までのブリッジ・セラピーとしてclass IIbの推奨度で紹介されている.しかしBASは小児例には推奨はされておらず肺移植までの待機中に増悪した場合,膜型人工肺が残された治療戦略となる.BASによる短絡の作成は,Eisenmenger症候群がIPAHに比してより安定した肺循環動態であり生命予後が良いことから発想されたものである.ワールド・シンポジウムでも取り上げられ,1980年代から重症PAH患者に対して施行されてきた.しかし,心房間短絡の自然閉鎖があり効果が一過性であることや全身の組織低酸素が生じることなどの理由から,その臨床的効果は決して高くはなかった.
外科的Pottsシャント
Pottsシャントは1946年にWillis J. Pottsによって報告された術式である30).1945年にBlalock–Taussigシャントが新しい術式として報告された時期であり,このシャント術が鎖骨下動脈を犠牲にする必要があることから,大動脈と左肺動脈を直接吻合する別の術式として紹介された.結局このPottsの術式はBlalock–Taussigシャントにとって代わることはなかったが,その後高度PAHを呈する症例に対する姑息的な治療法として再び注目されるようになる.前述のようにBASでは心房レベルの右左短絡によって全身のチアノーゼを来すのに対しPottsシャントでは下半身のみにチアノーゼを認めるため,脳や冠動脈への酸素供給が保たれるメリットがある(Table 2).小児例へのPottsシャントは2004年にBlancらにより報告されており,4歳と14歳の重症PAHに対し行われ劇的な回復が認められた31).2009年にはLabombardaらが,失神を繰り返していた2例の小児PAHに対しPottsシャントを行い症状の改善に成功している32).これら2つの報告によって,PottsシャントはPAHに対する新しい治療戦略として知られるようになり,2012年にBaruteauらによって小児IPAHの長期成績が報告された33).WHO肺高血圧症機能分類がIV度の2歳から17歳の8例に対し9 mm程度の短絡を作成し,2例が術後に肺高血圧クリーゼによって死亡したが,生存した6例はWHO機能分類のIまたはII度で維持できていた.その後,小児PAH症例に対するPottsシャントの症例報告があるが,いずれも良好な成績である34, 35).2018年のワールド・シンポジウムでも小児重症PAHへの治療として議論されており36),今後の新たな治療戦略になる可能性がある.
Table 2 Potts shunt | Balloon atrial septostomy | Potts shunt |
|---|
| Image | 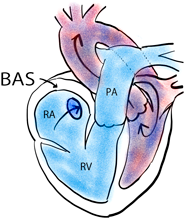 | 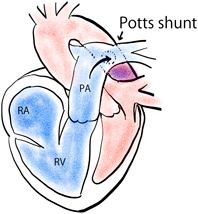 |
| Procedure | Creating a communication in the interatrial septum | Creating a communication between left pulmonary artery and descending aorta |
| Cyanosis | Intracardiac right-to-left shunt cause systemic desaturation | Postcardiac right-to-left shunt did not provoke arterial oxygen desaturation in coronary and cerebral circulations |
Pottsシャントの術式は下行大動脈と左肺動脈に直接吻合する方法であり,短絡血流の方向はコントロールすることはできない欠点が指摘されていたが,近年改良された術式が報告されている.下行大動脈と左肺動脈の間に距離がある症例に対しダクロン・グラフトを造設する術式や37),弁付きPottsシャントによって血流が肺動脈から大動脈方向にしか流れないような工夫をした術式などが考案されている38).弁付きPottsシャントは卵円孔と似た機序で,肺動脈圧が大動脈圧を超える場合のみ弁が開いて右左短絡が生じるようになっており,さらに改良された3弁付き導管を造設する術式も紹介されている39).
経皮的Pottsシャント
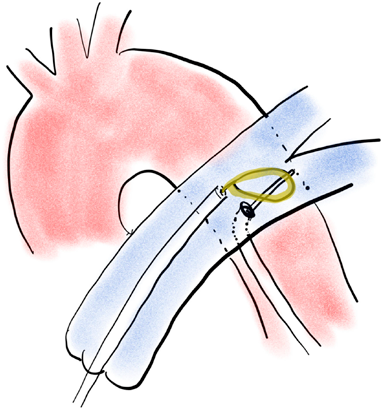
成人領域では外科的Pottsシャントは周術期で死亡率が高く,小児例に比して有効な治療戦略ではなかった.そこで,カテーテルによる経皮的Pottsシャントが試みられるようになった40).先行研究の動物モデルでの結果を参考に,2013年に18歳から44歳までの4例のIPAHに対して肺動脈大動脈の間にcoveredステントを留置する経皮的Pottsシャントが行われた41).大腿動脈に固定したロングシースからBrockenbrough針を出して穿通させ,この大動脈と肺動脈間に冠動脈用のバルーンカテーテルを通過させた後,このバルーンカテーテル先端から出してあったワイヤを予め左肺動脈に留置したスネアワイヤで掴みバルーンを拡張させる(Fig. 1).拡張後,coveredステントを短絡部位に置いて再度バルーン拡張させて左肺動脈と下行大動脈の間にシャントを作成する.この報告では,4例中3例が成功,1例は術中に血胸で死亡し,成功した3例中の1例も術後合併症によって死亡したが,2例は生存している.この結果からも,経皮的Pottsシャントは難易度が高く,限定された施設においてのみ施行されるべきである.
近年,再生療法が様々な領域において応用されている.勿論PAH患者に対する血管再生療法の臨床応用にはまだ時間がかかると考えられるが,この新しい治療には大きな期待が寄せられている.循環器領域の再生療法には,胚性幹細胞(embryonic stem cell, ESC),間葉系幹細胞(mesenchymal stem cell, MSC),血管内皮前駆細胞(endothelial progenitor cells, EPC)など様々な細胞が使用されてきた.一般に生体の幹細胞は多分化能を持ち,組織において必要とされる細胞に分化する.ESCは初期胚から分離される細胞で生体内の全ての細胞に分化する能力を持つことから多能性幹細胞(pluripotent stem cells, PSC)とも呼ばれ,人工的に作製したinduced PSCが今後の再生医療において重要な位置を占めている.PAHの実験モデルにおいて,幹細胞移植が肺の微小血管を再生することは報告されているが,細胞の培養増殖が難しい,生体内で増殖や分化が得られない,目標臓器で生着させられないなど臨床応用の前に解決すべき様々な課題が残されている.
間葉系幹細胞(MSC)
多分化能幹細胞と呼ばれるMSCはPSCとは異なり分化できる細胞が限定されているが,自己再生能を持つ細胞である.自己の骨髄から採取でき,その高い増殖能力から大量に培養することが可能である.MSCは血管内皮細胞や平滑筋細胞へ分化するだけでなく成長因子やケモカインを放出し,細胞増殖抑制,血管新生の促進,抗炎症作用など様々な作用によってPAHの病態を改善に導くことが可能と考えられている42).2006年以降MSCを用いたPAHに対する有効性が報告され,モノクロタリンのPAHモデルではMSCの経静脈的移植により平均肺動脈圧の低下だけでなく組織学的な改善を認めた43–46).このMSC治療の重大な問題点は肺塞栓のリスクが高いことで,動物モデルの40%程度が肺塞栓によって死亡している47).他にも投与する適切な細胞数や投与経路など,多くの課題があるが,今後可能性のある治療として期待されている.
血管内皮前駆細胞(EPC)
PAHの病態は,肺血管の内皮細胞障害から始まると考えられているが,これは血管損傷と修復のバランスが崩壊していることに基づいている.近年,EPCがPAHにおける血管修復に関与することがわかってきた48).EPCは単一の細胞ではなく様々な脈管形成にも関与するEPC様の細胞も存在し,循環血管内皮前駆細胞(circulating endothelial precursor)や循環血管新生細胞(circulating angiogenic cells)の名称でも呼ばれている.EPCはMSCから分化するが,末梢血中には極めて少数しか存在しないため,MSCと異なり採取と増殖は極めて難しい.2004年にPAHモデルにEPCの自家移植を行った報告では,肺動脈圧や肺血管抵抗,心拍出量が改善し,肺血管の組織学的な改善も得られている49).2008年には13例の小児IPAHに対し経静脈的なEPCの自家移植が行われ,重篤な有害事象は認めず肺循環動態やNYHA機能分類などが改善した50).また,先天性心疾患の小児PAH患者において,EPCの数が肺動脈圧に逆相関しておりEPC自体が肺循環動態に影響している可能性が指摘された51).2015年に行われたpulmonary hypertension and angiogenic cell therapy(PHACeT)の検討では,7例のIPAH患者に対し血管内皮由来のNO合成酵素を導入させたEPCを右心房内に経静脈的に投与したところ,3日目には肺循環動態の改善が得られた52).しかし,その治療効果は投与3か月後には消失し1例は死亡している.この結果からもMSC同様に臨床応用には時間が必要と考えられるが,PAHの病態からはEPC治療は有望と考えられ今後の研究成果が期待されている.
治療薬の開発において,ある薬剤が動物モデルで効果が得られた後にPAH患者に対する臨床試験が行われたものの,安全性や有効性に問題が生じて臨床応用が中止になることがあるが,ここではイマチニブの例について紹介する.
イマチニブ
蛋白質のリン酸化されるアミノ酸残基の1つがチロシンであり,これを特異的にリン酸化する酵素をチロシンキナーゼ(tyrosine kinase, TK)と呼ぶ.TKは細胞膜表面に存在する受容体型と細胞内の非受容体型に大別され,細胞の増殖,分化,アポトーシスに関わる.イマチニブはTK阻害薬として細胞のシグナル伝達を阻害し,細胞増殖を抑制,癌細胞などのアポトーシスを誘導する.慢性骨髄性白血病のがん細胞におけるTK活性を選択的に阻害するイマチニブにより予後の改善が認められている.
PAHの病態において,癌細胞と同様に無秩序な血管リモデングが生じているが,血小板由来増殖因子(platelet derived growth factor, PDGF)が肺動脈平滑筋細胞の過剰増殖に関与していることが知られている53).イマチニブはこのPDGF受容体にも抑制的に作用することで,新たなPAH治療薬として期待された.2005年,61歳のPAHの男性患者に対しイマチニブ200 mg/日の投与を行った症例報告で3か月後のNYHA機能分類,6分間歩行,および肺血管抵抗値の改善が得られ,その効果は6か月後も維持していた54).その後,第II相臨床試験を経てRCTであるIMPRES(imatinib in pulmonary arterial hypertension, a randomized efficacy study)が行われた55, 56).この試験では,既に肺血管拡張薬治療を受けている202例のPAH患者に対し,200~400 mgのイマチニブまたはプラセボが投与された.6分間歩行距離や肺循環動態に対してある程度の効果を示したが,予後の改善は得られず重篤な合併症として硬膜下血腫が認められたため,PAH治療に対する承認申請は取り下げられた.その後,他のTK阻害薬であるニロチニブやソラフェニブによるPAH治療が行われたが,逆にTK阻害薬自体がPAH発症に関与することがわかってきた57).
ダサチニブはTK阻害薬の1つであり,フィラデルフィア染色体陽性の急性リンパ球性白血病や慢性骨髄性白血病に対し投与される抗癌剤であり,薬剤性PAHの原因の1つとして考えられている.本薬剤によるPAH発症機序は明らかではないが,他のTK阻害薬よりもそのリスクが高いことが知られている58).一般にダサチニブによるPAHは可逆性であるが重篤例も報告されているため,血液腫瘍の治療中にはPAHのスクリーニングを行うことが推奨されている.また,ダサチニブ以外のTK阻害薬でもPAH発症への関連性が報告されている59).TK経路阻害による細胞の過剰増殖,肺血管リモデリングの抑制を行う発想は,新たなPAH治療薬として期待されたが,この経路の治療薬開発は難しいと考えられる.
まだ大規模な臨床研究は行われていないものの,PAHモデルでは有効性を見いだしている新たな治療経路は多く報告されており,有望なものをいくつか挙げて紹介する.
セロトニン阻害薬
セロトニン(5-hydroxytryptamine, 5HT)は脳の神経伝達物質でドーパミンやノルアドレナリンを制御し,その分泌が抑制されると鬱症状などの精神症状を引き起こす.5-HTは必須アミノ酸であるトリプトファンから産生され,5-HT受容体に結合し活性化する.この受容体には1~7型あり,さらにサブタイプにもわかれているが5-HT1B受容体がPAHの病態に関与する60).この5-HT1B受容体はPAH患者の肺血管において多く発現しており,5-HTが肺血管平滑筋細胞から5-HT輸送体(serotonin transporter, SERT)を介して5-HT1B受容体に運ばれて結合すると,肺血管平滑筋の収縮や細胞増殖,繊維化を引き起こす.この作用は5-HT1B受容体を介した酸化ストレスに伴う活性酸素種によってさらに増強される61).
鬱病に対して選択的セロトニン再取り込み阻害薬(selective serotonin reuptake inhibitor, SSRI)が用いられているが,SSRIはSERTの抑制を行うことで脳内神経伝達物質としての5-HTの再取り込みを阻害し,脳内での5-HT作用は増強され抗鬱作用を示す.一方,PAHモデルではSERTが過剰発現しており,このSSRIによってPAH進行が改善する可能性が示唆されている62, 63).しかし,妊娠第3期の後期に母体にSSRIが投与されると,新生児遷延性肺高血圧症の発症リスクが高くなることや,PAH患者でSSRIが投与されている場合は予後不良になる症例が多いこと,SSRIの使用例にPAH発症が多いとする報告もあり,SSRIがPAHの発症や進行に対して抑制的に作用するのか促進的に作用するのかは不明なところが多い64–66).現状,選択的5-HT1B受容体阻害薬の臨床的な評価が不十分であり,更なる検討が必要と考えられる.
グリチルリチン
HMGB1(high mobility group box-1)は,DNA構造の維持や機能発現に重要な生体内の核内タンパクである.HMGB1は細胞の壊死や活性化に伴い核から細胞外に放出され,Toll様受容体–4などを介して炎症性サイトカインの発現を誘導することから,“ダメージ関連分子パターン”と考えられている.グリチルリチンはHMGB1と結合して,その作用を中和する作用がある67).生薬の甘草に含まれるグリチルリチンは,砂糖より甘味の強い物質であり強力ネオミノファーゲンシー®の主成分として肝機能障害に対して使用されている.作用機序は不明なところが多いが主に抗炎症作用によるものであり,マクロファージの活性化を抑え,炎症性ケミカルメディエーター放出を抑制する.また,内因性のインターフェロンγを産生し免疫応答を増強させることで,ウイルスの増殖抑制に働くことも知られている.
PAHの肺血管のリモデリングにおいて,炎症が大きく関わっていると考えられている.PAHでは様々な血清中のケモカイン,サイトカインの上昇が認められ,マクロファージ,リンパ球などが血管内皮細胞に浸潤している68, 69).さらに,低酸素によるPAHモデルやIPAH患者ではHMGB1が上昇していることが報告されており,PAH患者の病態の進行においてこのHMGB1による炎症が重要な役割をもっていることがわかってきた70–72).PAHモデルに対して,グリチルリチン投与した検討では,HMGB1の発現抑制に加え肺循環動態が改善した73, 74).この結果から,PAHの病態における“炎症”を抑制することは,疾患の進行に影響することが考えられ,今後新しい治療標的になることが期待される.
ラノラジン
ラノラジンは2006年に慢性狭心症の治療薬として承認された薬剤で,その薬理作用は十分解明されていないものの,アデノシン三リン酸の産生促進による心筋血流の増加をきたし心保護作用を示すと考えられている75).心拍数の減少や血圧低下を来さずに心筋酸素消費量の改善させることができることが特徴である.また,Na channelの感受性を低下させ抗不整脈作用を併せ持つが,副作用としてQT延長を来すことが知られている76).ラノラジンによるPAHモデルへの治療効果を検討した報告では,右室駆出率と右室リモデリングの改善が得られた77–79).その後,12例のPAH患者に対して単施設におけるラノラジン治療のRCTが行われたが,12週間の投与によって明らかな効果が得られなかった80).また,11例のPAH患者に対するラノラジン治療の前向き臨床試験の検討では,NYHA機能分類の改善と右室容積の縮小や右室機能の改善が得られたが,明らかな肺循環動態の改善は得られなかった81).本薬剤の薬理作用からはPAHへの治療効果が期待されたが,今後有効な治療となり得るかは更なる検討が必要である.
エストロゲン
PAHにおける女性ホルモンのエストロゲンの関与については長く議論されてきた.一般的に小児期PAHでは発症率に性差がないのに対し,成人領域では思春期を超えると女性の方が男性に比して2~4倍もの発症リスクがある82).しかし,PAHの重症度は女性よりも男性の方が高く予後においても男性の方が不良である83).このようにPAH発症には女性ホルモンの影響が疑われるが,重症度や予後とは解離した結果を示すことから“エストロゲン・パラドックス”と呼ばれており,その病態への関与についてはまだ十分解明されていない.
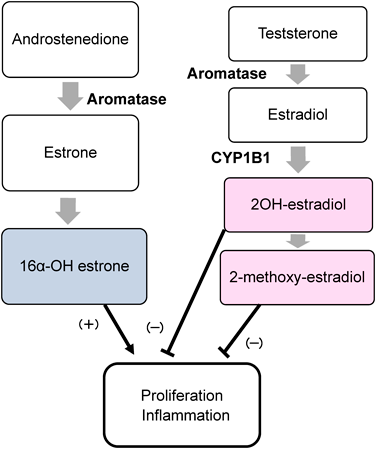
エストロゲンはプロスタサイクリンやNO分泌を促進しエンドセリン1の発現を抑制するが,一方で肺血管の平滑筋においてその増殖に関与すると考えられている84).このエストロゲンの肺血管における相反する作用を示す機序は,エストロンとエストラジオールの異なる作用によって説明される85).エストラジオールの代謝産物である2-methoxy-estradiol, 2-OH-estradiolは増殖抑制と抗炎症作用を示し,一方でエストロンの代謝産物の16α-OH-estradiolは増殖促進と炎症作用を亢進させる(Fig. 2).チトクロームP450分子種の一つとして同定されたCYP1B1(cytochrome P450 1B1)は乳癌の発症に関与する酵素として知られているが,健常ではエストラジオールから2-methoxy-estradiolに分解する酵素である.このCYP1B1を抑制すると,エストロンから16α-OH-estradiolへの変換が相対的に増加し,PAHにおいて病態を悪化させる方向に働く86).また,BMPR2(bone morphogenetic protein receptor type II)遺伝子変異を持つ症候性PAHの女性例におけるCYP1B1の発現は,無症候性のBMPR2遺伝子変異の症例に比して有意に低下していたが男性ではこの傾向は認められていない87).さらにBMPR2遺伝子変異を持つ無症候性のキャリアにおいても,遺伝子変異のない症例に比して2-OH-estradiol/16α-OH-estrone比が著しく低下していた88).これらの結果から,女性のPAHではCYP1B1の発現の低下によって2-methoxy-estradiolよりも16α-OH-estradiolが増加しており,一方男性では,この代謝系によるPAHの病態への関与は認められないことが考えられる.このエストロゲンを標的としたPAH治療の検討がされており,PAHのモデルに対するエストラジオール受容体の選択的アゴニストの投与により肺循環動態の改善を認めたことが報告されている89–91).また,生体内で男性ホルモン(アンドロステンジオン,テストステロン)から女性ホルモン(エストロン,エストラジオール)に分解する酵素のアロマターゼが,閉経後の女性の肺動脈平滑筋に多く発現しており,肺高血圧の進展に関与している可能性が示唆された92).このアロマターゼの抑制によりPAHの改善が得られる可能性があり,アロマターゼ阻害薬(アナストロゾール)によるRCTが行われた93).18例のPAH患者(女性9例)に対しアナストロゾール治療群とプラセボ群で比較検討されたが,治療開始3か月後に治療群では6分間歩行距離の延長を認めたものの,NTproBNP値やNYHA機能分類は改善がなかった.アナストロゾールがエストロンとエストラジオールの経路を同時に抑制したことで,臨床的な改善が得られなかった可能性はあるが,まだこの経路への治療は難しいことが考えられる.このようにPAHの病態においてエストロゲンの関与は明らかであるものの,動物モデルとは異なり実際の患者への治療においては未解決なものが多く残されている.また生体内のホルモンバランスを崩す可能性もあり問題点は多いと考えられる.長くこの分野への取り組みはなされているが治療への応用には到達できておらず,治療薬開発にはまだ時間がかかりそうである.