2018年2月にNiceで開催された6th World Symposium on Pulmonary Hypertension(WSPH)での検討内容が,同年12月に成文化された.このなかでの最大のトピックスは,何と言っても肺高血圧(pulmonary hypertension: PH)の定義が,安静時の平均肺動脈圧mean pulmonary arterial pressure: mPAP「25 mmHg以上」から「20 mmHgを超えるもの」へと変更されたことであろう(Table 1)1, 2).この変更の根拠として,健常者のmPAPが14.0±3.3 mmHgであったこと3),mPAPが21~24 mmHgの場合,20 mmHg以下の場合と比較して,運動耐容能が低く,かつ入院率および死亡率が上昇したと報告されたことなどが挙げられる4, 5).ただし,mPAPは心拍出量cardiac output: COや肺動脈楔入圧pulmonary artery wedge pressure: PAWPの影響を受けることから,今回この6th WSPHでは前毛細血管性肺高血圧pre-capillary PHの定義として,上記のmPAP>20 mmHgに加えて,肺血管抵抗pulmonary vascular resistance: PVR≧3Wood Unitsが挙げられ,発症原因とは無関係にこの定義を使用することとされた.mPAP 21~24 mmHgかつPVR≧3Wood Unitsの患者に対するPAH治療の意義については現時点ではまだ不明であり,今後検討されるべき課題である.
Table 1 New definition of pulmonary hypertension| Definition | Characteristics | Clinical groups |
|---|
| Pre-capillary PH | mPAP>20 mmHg | group 1: PAH |
| PAWP≦15 mmHg | group 3: PH due to lung diseases and/or hypoxia |
| PVR≧3WU | group 4: PH due to pulmonary artery obstructions |
| | group 5: PH with unclear and/or multifactorial mechanisms |
| Isolated post-capillary PH | mPAP>20 mmHg | group 2: PH due to left heart disease |
| PAWP>15 mmHg | group 5: PH with unclear and/or multifactorial mechanisms |
| PVR<3WU | |
| Combined pre- and post-capillary PH | mPAP>20 mmHg | group 2: PH due to left heart disease |
| PAWP>15 mmHg | group 5: PH with unclear and/or multifactorial mechanisms |
| PVR≧3WU | |
PH: pulmonary hypertension; PAWP: pulmonary arterial wedge pressure; PVR: pulmonary vascular resistance; WU: Wood Units.
[Adapted from Ref.2)] |
さらに,生後3か月以上の小児においても,前述の成人PHに倣い,mPAP 20 mmHgを超えるものをPHと定義することとなった.PVRについては既にこれまでの小児PH評価に取り入れられている,体表面積で補正したPVR Index (PVRI)≧3Wood Units·m2を使用することがあらためて推奨されている.成人例と同様,mPAP 21~24 mmHgの小児患者への対応については今後さらに検討を重ねる必要がある.
PHの臨床分類については前回の5群が踏襲されたものの,小分類についてはいくつかの改定がなされた(Table 2).
Table 2 Updated clinical classification of pulmonary hypertension| Group 1: PAH |
| 1.1 Idiopathic PAH |
| 1.2 Heritable PAH |
| 1.3 Drug- and toxin-induced PAH |
| 1.4 PAH associated with: |
| 1.4.1 Connective tissue disease |
| 1.4.2 HIV infection |
| 1.4.3 Portal hypertension |
| 1.4.4 Congenital heart disease |
| 1.4.5 Schistosomiasis |
| 1.5 PAH long-term responders to calcium channel blockers |
| 1.6 PAH with overt features of venous/capillaries (PVOD/PCH) involvement |
| 1.7 Persistent PH of the newborn syndrome |
| Group 2: PH due to left heart disease |
| 2.1 PH due to heart failure with preserved LVEF |
| 2.2 PH due to heart failure with reduced LVEF |
| 2.3 Valvular heart disease |
| 2.4 Congenital/acquired cardiovascular conditions leading to post-capillary PH |
| Group 3: PH due to lung diseases and/or hypoxia |
| 3.1 Obstructive lung disease |
| 3.2 Restrictive lung disease |
| 3.3 Other lung disease with mixed restrictive/obstructive pattern |
| 3.4 Hypoxia without lung disease |
| 3.5 Developmental lung disorders |
| Group 4: PH due to pulmonary artery obstructions |
| 4.1 Chronic thromboembolic PH |
| 4.2 Other pulmonary artery obstructions |
| Group 5: PH with unclear and/or multifactorial mechanisms |
| 5.1 Haematological disorders |
| 5.2 Systemic and metabolic disorders |
| 5.3 Others |
| 5.4 Complex congenital heart disease |
PAH: pulmonary arterial hypertension; PH: pulmonary hypertension; PVOD: pulmonary veno-occlusive disease; PCH: pulmonary capillary haemangiomatosis; LVEF: left ventricular ejection fraction.
[Adapted from Ref.2)] |
1群「肺動脈性肺高血圧症pulmonary arterial hypertension: PAH」については,1.5群として新たに「カルシウム拮抗薬に長期反応を示すPAH(PAH long-term responders to calcium channel blockers)」が加えられた.以前から,肺血管拡張試験に良好な反応を示したPAH患者群において,カルシウム拮抗薬の長期投与が生存率を向上させたとの報告が散見されている6, 7).PAHの主要な原因が肺小動脈リモデリングであることには疑いはないが,一方で肺血管収縮もPAHにおいて重要な役割を担っていることが再評価されたものと考えられる.ただし,肺血管拡張試験の対象としてはIPAH, HPAHおよびdrug-induced PAHの患者が望ましく,他のタイプのPH, PAHでは同試験は勧められないと警告されている.カルシウム拮抗薬を開始した場合にはその3~6か月後に血行動態の改善を確認すること,改善が見られなかった場合には肺血管拡張試験陰性のPAH患者に対する治療戦略に移行することが推奨されている.移行した症例のうち,少なくとも1年間,カルシウム拮抗薬のみで血行動態が改善していた患者に対しては,combination therapyではなく肺血管拡張薬1剤の導入が望ましいとされている8).肺血管拡張試験の陽性例は少ないものの,小児においては成人よりも陽性例が比較的多いとされており,小児IPAH症例の8~15%程度が陽性となるのではないかと推定されている9).なお,肺血管拡張試験の結果が正確に解釈されていない場合があり,注意が必要である.例えば小児IPAH/HPAHにおいて,主治医が肺血管拡張試験陽性と臨床的にみなした症例が37%であった際に,実際にSitbon criteria(mPAPが10 mmHg以上低下し,その絶対値が40 mmHg未満となり,かつ心拍出量が不変または増加した場合に陽性とする.6th WSPHにおいても,小児IPAH/HPAHにおける肺血管拡張試験の基準としてSitbon criteriaを使用するよう推奨された)を満たしたのは15%と解離が見られたとの報告もある10).その原因については,患者の選択,同試験に用いる肺血管拡張物質の選択,反応があったとみなす基準などを含めて作成されている現在のPAHガイドラインに,実際の小児IPAH/HPAH診療が即しておらず,主治医によって対応が異なっていることが挙げられている.また,前述の海外からの報告で使用されているカルシウム拮抗薬の用量が非常に多く,同量で投与した場合の体血圧低下を懸念して,本邦では実際には,PAHに対してカルシウム拮抗薬を単独で使用することは非常に稀である.しかしながら,肺血管拡張試験が明らかに陽性であった症例ではその使用は検討すべきと考えられる.
前回5th WSPHで定められた臨床分類で1′群とされていた肺静脈閉塞性疾患pulmonary veno-occlusive disease: PVOD/肺毛細血管腫症pulmonary capillary haemangiomatosis: PCH,新生児遷延性肺高血圧症候群persistent pulmonary hypertension of the newborn syndrome: PPHNは,それぞれ1.6群,1.7群へと変更された.このうち,PVOD/PCHが,PAH with overt features of venous/capillaries(PVOD/PCH)involvement,すなわち「PVOD/PCHの明確な特徴を有するPAH」と変更されたことは,前述のPH, PAHの定義変更と同等の,特筆すべきポイントであると個人的にはとらえている(詳細は3章で述べる).全身性硬化症のようなPAHを惹起する疾患において肺静脈や肺毛細血管にも異常が見られるとの報告があること,PAHとPVOD/PCHの臨床像や血行動態が非常に類似していること,PVOD/PCHの原因遺伝子であるEIF2AK4変異を持つ患者で肺動脈リモデリングが認められたこと11),逆にPAHの主要な原因遺伝子であるBMPR2変異を持つ患者において肺静脈の筋性リモデリングが認められたことが12),今回の再分類の根拠となっている.
5群は「PH with unclear and/or multifactorial mechanisms原因不明/複合要因による肺高血圧症」に改められ,さらに5.4群としてcomplex congenital heart disease,複雑先天性心疾患が独立して明記された.この5.4群は区域性PH (Segmental PH),単心室およびその類縁疾患,そしてScimitar症候群からなる(Table 3)9).特にFontan循環となっている単心室およびその類縁疾患については,慢性的に非拍動的な肺循環となっており,他の二次性PHを引き起こす疾患とは明らかに病態が異なる.PHの診断基準であるmPAP>20 mmHgを満たすことも通常はない.それゆえにこれまでの臨床分類からはFontan循環は除外されていたが,その肺血管病変が生存率に大きく関わることを重視し,今回5群PHとして分類された.そのFontan循環が肺血管病変を惹起するメカニズムなどについては今後の検討課題である.
Table 3 Group 5.4 Complex congenital heart disease| Segmental pulmonary hypertension |
| Isolated pulmonary artery of ductal origin |
| Absent pulmonary artery |
| Pulmonary atresia with ventricular septal defect and major aorto-pulmonary collateral arteries |
| Hemitruncus |
| Other |
| Single ventricle |
| Unoperated |
| Operated |
| Scimitar syndrome |
| [Adapted from Ref.9)] |
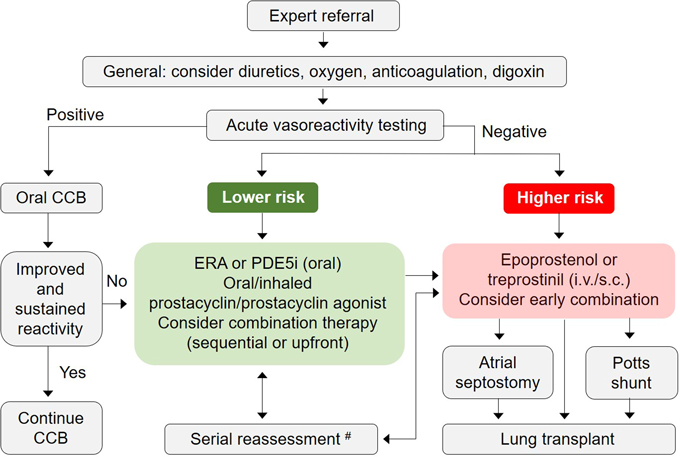
小児PAHの治療アルゴリズムについては,成人PAHを対象とした各種臨床研究も参考として改変された(Fig. 1).肺血管拡張試験で陽性だった場合は経口カルシウム拮抗薬を投与し,陰性だった場合はTable 4の各リスク因子を照らし合わせ,その重症度を見極めて治療方針を判断することとなる.このリスク分類は6th WSPH成人タスクフォースでは,2015年の欧州心臓病学会/欧州呼吸器学会の肺高血圧症ガイドラインをそのまま踏襲したものとなっている8, 13).小児タスクフォースでのリスク分類の主な変更点としては,失神の有無の項目が削除されたこと,6分間歩行距離(6歳以上の場合)が追加されたこと,心臓超音波の項目に三尖弁輪収縮期移動距離(tricuspid annular plane systolic excursion: TAPSE)および右室面積変化率(right ventricular fractional area change: RVFAC)の低下が加えられたこと,血行動態の項目に混合静脈血酸素飽和度(systemic venous oxygen saturation: SvO2)および肺動脈コンプライアンス係数(pulmonary arterial compliance index: PACI.肺血流量を体表面積および心拍数で除し,さらに(収縮期肺動脈圧–拡張期肺動脈圧)で除して求められる)が追加されたことが挙げられる(Table 4).各項目の変更理由やその思考過程について,現在のところ小児タスクフォースからは詳細には言及されていないが,PACIについては,2013年のDouwesらによる,難治性の小児PAHにおいてPACIがNYHA分類および予後とよく相関したとの報告を重視したものと考えられる14).
Table 4 Determining factors of paediatric idiopathic/heritable pulmonary arterial hypertension risk| Lower risk | Determining factors | Higher risk |
|---|
| No | Clinical evidence of RV failure | Yes |
| No | Progression of symptoms | Yes |
| >350 | 6MWT (>6 years old) m | <350 |
| Normal | Growth | Failure to thrive |
| WHO FC | III, IV |
| Minimally elevated | Serum BNP/NT-proBNP | Significantly elevated |
| Rising level |
| Echocardiography | RA/RV enlargement |
| Reduced LV size |
| Increased RV/LV ratio |
| Reduced TAPSE |
| Low RV FAC |
| Pericardial effusion |
| Systemic CI >3.0 L·min−1·m−2 | Haemodynamics | Systemic CI <2.5 L·min−1·m−2 |
| Systemic venous saturation>65% | mRAP >10 mmHg |
| Acute vasoreactivity | PVRI >20 WU·m2 |
| Systemic venous saturation<60% |
| PACI <0.85 mL·mmHg−1·m−2 |
RV: right ventricle; 6MWT: 6-min walk test; WHO: World Health Organization; FC: Functional Class; BNP: brain natriuretic peptide; NT-proBNP: N-terminal pro-BNP; RA: right atrium; LV: left ventricle; FAC: fractional area change; TAPSE: tricuspid annular plane systolic excursion; CI: cardiac index; mRAP: mean right atrial pressure; PVRI: pulmonary vascular resistance index; WU: Wood Units; PACI: pulmonary arterial compliance index {[stroke volume/(systolic pulmonary artery pressure-diastolic pulmonary artery pressure)]/body surface area}.
[Adapted from Ref. 9)] |
今回の小児PAHの治療アルゴリズム(Fig. 1)について注目すべきは,肺血管拡張試験の結果が陰性かつ低リスク群にあたる症例については経口肺血管拡張薬1剤での治療開始を推奨しながらも,早期のcombination therapy(sequentialもしくはup-front)が有用である可能性を明記したことである.しかし,どのような症例でどちらを選ぶべきかは不明であり,また,sequentialとup-front combination therapyの比較も特になされていない.一方,成人PAHの治療アルゴリズムでは,低リスク~中等度リスク群に対しては,当初から複数の経口肺血管拡張薬を導入することを基本としながらも,高齢者やヒト免疫不全ウイルス関連PAH患者,非常に軽症のPAH患者など,monotherapyのほうが望ましい可能性がある病態について列挙している(Table 5)8).本来は小児PAHにおいても,monotherapyが望ましい,つまりはcombination therapyを避けるべき群について明記すべきだったのではないかと筆者としては考えている.それは既報や自験例から,Table 5の事項のなかでも特に,小児においても,PVOD/PCHなどの合併が疑われる治療抵抗性PAH症例への対応を非常に懸念しているためである.
Table 5 Potential role for initial monotherapy in specific pulmonary arterial hypertension (PAH) subsets| ・IPAH, HPAH and drug-induced PAH patient responders to acute vasoreactivity tests and with WHO FC I/II and sustained haemodynamic improvement (same or better than achieved in the acute test) after at least 1 year on CCBs only |
| ・Long-term-treated historical PAH patients with monotherapy (>5–10 years) stable with low-risk profile |
| ・IPAH patients >75 years old with multiple risk factors for heart failure with preserved LVEF (high blood pressure, diabetes mellitus, coronary artery disease, atrial fibrillation, obesity) |
| ・PAH patients with suspicion or high probability of pulmonary veno-occlusive disease or pulmonary capillary haemangiomatosis |
| ・Patients with PAH associated with HIV infection or portal hypertension or uncorrected congenital heart disease, as they were not included in RCTs of initial combination therapy |
| ・PAH patients with very mild disease (e.g. WHO FC I, PVR 3–4 WU, mPAP <30 mmHg, normal right ventricle at echocardiography) |
| ・Combination therapy unavailable or contraindicated (e.g. severe liver disease) |
IPAH: idiopathic PAH; HPAH: heritable PAH; CCB: calcium channel blocker; PAP: pulmonary arterial pressure; PVR: pulmonary vascular resistance; LVEF: left ventricular ejection fraction; RCT: randomised controlled trial; WHO: World Health Organization; FC: Functional Class; WU: Wood Units; mPAP: mean PAP.
[Adapted from Ref.10)] |
次項以降では1.2群Heritable PAH: HPAHと,1.6群PAH with overt features of venous/capillaries(PVOD/PCH)involvementに主に焦点を当てる.さらに後者に関連して小児重症PAHにおける治療戦略について再考することとする.
いわゆる二次性PAHを除いたPAHの約10%で家族歴が認められ,その発症に遺伝学的要因が関与している可能性は30年以上前から指摘されていた15).2000年に,HPAHの疾患原因遺伝子として形質転換成長因子(transforming growth factor-beta, TGF-β)スーパーファミリーに属する骨形成タンパク(bone morphogenetic protein, BMP)シグナル伝達経路内の2型骨形成タンパク受容体(bone morphogenetic protein receptor 2: BMPR2)がはじめて報告され16, 17),その後同じくTGF-β/BMPシグナル伝達経路に属する1型アクチビン受容体様キナーゼ(activin receptor-like kinase 1: ACVRL1),エンドグリン(endoglin: ENG),転写調節因子SMAD8(SMAD9)が疾患原因遺伝子として相次いで報告され18–21),さらに2012年にカベオリン-1(caveolin-1, CAV1)が,2013年にはKCNK3が報告された22, 23).CAV1は,細胞膜上で陥凹構造を示すcaveolaeの形成に必須であり,またeNOS(endothelial NO synthase)の放出を介して血管形成に関与すること.さらに,TGF-βスーパーファミリーを含む多くの細胞表面レセプターがcaveolae内にあり,caveolae形成がTGF-βシグナル伝達経路に関与するとの報告もあることから,CAV1変異がTGF-βシグナル伝達経路に影響を与えている可能性が示唆される22).KCNK3は4回膜貫通型カリウムチャネルのひとつであり,ヒト肺動脈平滑筋細胞に発現し,肺動脈の緊張性に関与していると考えられている24).また,カリウムチャネルがもたらす膜電位および細胞体積の変化は細胞周期の進行に必須と考えられていることから,KCNK3変異が肺動脈平滑筋細胞の増殖に関わっている可能性がある25).
前回5th WSPHの際には,1.2群Heritable PAHの原因遺伝子としてこれらの6遺伝子,BMPR2, ACVRL1, ENG, SMAD8(SMAD9), CAV1, KCNK3が明記されたが26),今回の6th WSPHではそれぞれのエビデンスレベルに基づき17遺伝子が疾患原因遺伝子として挙げられた(Table 6)27).このうちのひとつであるTBX4は,2013年に膝蓋骨の無形成/低形成,寛骨の骨化異常,第1~2趾間の広い間隙等を特徴とし,常染色体優性遺伝形式をとる小膝蓋骨症候群(small patella syndrome, SPS)28)の原因遺伝子として知られており,PAH発症にも関与しているのではないかと報告されていた29).その後,成人PAHとは異なり,小児PAHにおいてはこのTBX4変異,およびACVRL1変異が非常に多く同定されることが明らかになった29–32).また,上記の17遺伝子以外にも最近,ATP依存性カリウムチャネルであり,高インスリン血症の原因遺伝子としても知られている,ABCC8遺伝子変異が12名のPAH患者で同定され,さらに,高インスリン血症の治療薬であるジアゾキサイドがこの変異カリウムチャネルの機能を改善させたとの報告がなされている33).
Table 6 Classification of pulmonary arterial hypertension genes according to level of evidence that they play a causal role in the disease| Higher level of evidence | Lower level of evidence |
|---|
| BMPR2; EIF2AK4; TBX4; ATP13A3; GDF2; SOX17; AQP1; ACVRL1; SMAD9; ENG; KCNK3; CAV1 | SMAD4; SMAD1; KLF2; BMPR1B; KCNA5 |
Evidence includes de novo mutation, cosegregation studies, association with replication and functional studies.
[Adapted from Ref. 25)] |
これらの疾患原因遺伝子の同定により,分子生物学的側面からのPAHの病態についての理解が急速に進歩したのは事実である.しかし,PAHに限ったことではないが,エクソーム解析の結果のみで新規疾患遺伝子候補を同定するのは危険である.エクソーム解析後の候補遺伝子の絞り込みの手法にはいまだ統一されたものが存在しない.そして,エクソーム解析で病的意義が不明であるvariant(variant of unknown significance: VUS)を検出した際の対応は千差万別である.VUSについては患者やその家族はもちろん,主治医にも一切伝えないと取り決めている研究施設もあれば,患者およびその家族に伝えた上で独自に機能解析を進める施設や,残念ながら追加検討を一切行わずに,新規変異としての公表を試みようとする施設も存在する.また,BMPR2のようにPAHの疾患原因遺伝子として確定している遺伝子であっても,その遺伝子内の新規variantの全てが病的とは限らないため,このような場合も一例一例慎重に吟味する必要がある.遺伝学的検査の受託施設はもちろんのこと,検査を依頼する側の施設においても遺伝学的検査結果を鵜呑みにはせず,米国臨床遺伝学会American College of Medical Geneticsおよび分子病理学会Association for Molecular Pathologyのガイドラインを参照することを推奨したい34).
HPAHの約20~30%,およびIPAHの約60~90%では上記の遺伝子変異は見つかっていない30, 35, 36).また,各々の遺伝子変異とPAHの臨床像との関係は十分に明らかにはされておらず,特に小児PAHに関する解析は皆無であった.そこで,筆者らは小児期発症PAH症例について,遺伝子変異の有無と臨床像の関係をはじめて検討した37).小児期発症PAH54症例を,18症例のBMPR2変異群,7症例のACVRL1変異群,既知のBMPシグナル伝達経路内の遺伝子変異を認めない29症例(以下非変異群)の3群へと分類し,臨床像を検討したところ,非変異群では5年生存率,10年生存率ともに90%であったのに対して,BMPR2変異群では55%,37%と明らかに予後不良であった(Fig. 2).症例数が少ないために有意差はなかったものの,ACVRL1変異群もイベント発生リスクが非変異群の約5倍と,予後不良な傾向にあることが示唆された.BMPR2変異群およびACVRL1変異群については早期のエポプロステノール持続静注の導入や肺移植を含めた,より積極的な治療の選択を検討する必要があると考えられる.
成人発症PAHにおける遺伝子変異と転帰に関する報告は複数存在するが,各々の結果には解離がある38–41).筆者らの研究は小児期発症例のみに限定して検討したため,BMPR2変異群におけるPAH進行が非常に速く,予後が不良なために成人に至る例が少なく,成人例の検討には含まれにくい可能性が考えられる.2016年にEvansらにより報告された全世代のPAH症例1,550例を対象としたmeta-analysisによると,BMPR2遺伝子変異を有し,かつ若年時に発症したPAH患者ではより重症であり,死亡もしくは肺移植に至るリスクが高いことが示された.成人期発症例が中心となっている報告ではあるが,この報告は我々の研究結果を支持しうるものである42).同じく2016年にLevyらから報告された小児PAHでの検討では,原因遺伝子の有無と臨床像の関係は明らかにはならなかったが,BMPR2 5例,ACVRL1 4例,TBX4 3例等と,各々の症例数が少ないことが原因ではないかと考えられる31).真に臨床現場で役立つ意義を見いだすには,小児PAHに関する国際的な症例データベースの構築が望ましい.
PAH症例における遺伝学的検査の適応については様々な検討がなされてきたが,最近は全例で遺伝学的検査を考慮し,患者に助言すべきとの意見が増加している43, 44).特にフランスでは,2003年以降,PAH患者全員(薬剤性および膠原病性等の二次性PAHを含む)に,National referral centreにおける遺伝カウンセリングとBMPR2変異検索の実施を提示している44).6th WSPHでも,IPAH, familial PAH, anorexigen-associated PAH, PVOD/PCH, CHD-PAHの患者に対しては,それが遺伝性疾患である可能性,および患者の他の家族がPAHを起こす変異を持っている可能性があることを知らせる“medicolegal duty”があるとしている27).
小児PAH患者については,European Paediatric Pulmonary Vascular Disease(PVD)Networkが「原因不明の小児PAH患者において,BMPR2などの原因遺伝子検索は,機序の解明,予後の予測,他の家族の発症リスクを知るために有用となりうる」としており45),American Heart Association/American Thoracic Society(AHA/ATS)も同様の見解を出していた46).今回の6th WSPH小児タスクフォースでは精神面での影響を考慮すると,小児PH症例においては,遺伝学的検査はまだroutineで行うべきものではないとされた.また,遺伝学的検査の前後で遺伝カウンセリングを実施すべきと提案された9).
筆者個人としては,先に示したように,遺伝子変異の有無とその種類が予後に大きく影響する可能性があることから,少なくとも小児期発症PAH患者については全例に対して,遺伝学的検査の意義について十分説明するべきであると考えている.患者もしくは保護者の同意が得られた場合には遺伝学的検査を行う.家系内発症が認知されていたにもかかわらず,運動時に突然死し,剖検によりはじめてPAHと判明した小児例に関わった経験からも,原因遺伝子変異が見つかった患者の家族においても同意が得られた場合は積極的に遺伝学的検査を行うのが望ましいと考えている.さらにEvansらの報告42)からは,成人期発症PAH患者においても,少なくともBMPR2変異の有無は可能な限り全例で検索するのが望ましいであろう.
3. PAH with overt features of venous/capillaries(PVOD/PCH)involvement
PVODは末梢肺静脈の内膜の肥厚や線維化が進行することでPHを惹起する,稀で予後不良な疾患である47, 48).PVODの原因として,これまでにはウイルス感染や骨髄移植,アルキル化薬などの影響が指摘されている47).PCHは肺胞壁や気管壁,胸膜まで広がる過剰な毛細管様微小血管の増生や多層化によってPHを惹き起こす,やはり稀で致死的な疾患である49).
臨床的には両者ともPAHとの鑑別が困難であることが多いとされてきたが,1章で述べたように,6th WSPHにおいては1.6群PAH with overt features of venous/capillaries(PVOD/PCH)involvement,「PVOD/PCHの明確な特徴を有するPAH」としてまとめられた.
2014年に,家族性PVOD 13家系および孤発性PVOD 5例においてEIF2AK4が原因遺伝子として同定された50).さらに家族性PCHの1家系および孤発性PCHの2例において,同じくEIF2AK4が原因遺伝子として同定された51).これまでにPAHで同定されている原因遺伝子とは異なり,この変異は常染色体劣性遺伝形式をとる.PVODとPCHの2疾患は,これまで別々の疾患としてとらえられてきたが,現在では同一のスペクトラムにあると考えられている.PVOD/PCHの20~25%でEIF2AK4変異が検出される50, 51).また,臨床的PAHの約10%は病理学的にPVODであったとの報告がある47, 52).PVOD/PCHにおけるEIF2AK4変異同定の文献報告を受け,筆者も,これまでに既知の原因遺伝子変異が同定されていなかったPAH 74症例において,EIF2AK4変異を検索したが同変異は検出されなかった.しかし最近,臨床的にIPAH/HPAHとされた864名のうち,9名(1.0%)で,PVOD/PCHの原因遺伝子であるEIF2AK4変異が同定され,このうち1名では肺動脈内膜線維化と肺静脈の線維化が認められ,ほぼ完全に肺静脈が閉塞していた部位も見られたとの報告がなされた53).これらの既報を踏まえ,今回の6th WSPHにおいてPVOD/PCHはPAH with overt features of venous/capillaries(PVOD/PCH)involvementとして新規に分類されることとなった.
実際,臨床的にはPVOD/PCHの診断確定は特に小児では非常に困難である.これまで筆者が関わってきたなかでも,難治性の小児期発症IPAHとして診療されていたが,病理解剖でPVOD/PCHと診断された症例が存在する.肺生検は最も確実な診断方法であるが,出血や気胸,空気塞栓などのリスクがあり,推奨はされていない.また,病変の主座が肺動脈の末梢側にあるPH患者において,適切な部位から検体を採取するのは困難である.6th WSPHでは,肺拡散能力(diffusing capacity of lung for carbon monoxide: DLco)の著明な低下(予測値の60%未満)もしくは運動時の重度の低酸素血症はPVOD/PCHを示唆するものとして注意喚起しているが54),小児では呼吸機能や運動時の低酸素血症を適切に評価できる症例は限定される.高分解能CT(high-resolution computed tomography; HRCT)で小葉間隔壁の肥厚やすりガラス様陰影なども有用な所見とされているが,PVOD/PCHの全例でそのような所見が認められるわけではない.
PVOD/PCHに対しては肺移植が唯一の治療法とされている一方で,適応外使用となるが,PVODに対して,PDGF受容体のリン酸化を阻害するimatinibが有効であるとの報告が散見されている55, 56).現在のところ小児に対する使用例の報告はない.
4. 小児の重症“肺動脈性”肺高血圧症における治療戦略
小児期発症PAHとPVOD/PCHがoverlapしている症例は,実際どの程度存在しているのであろうか?また,それらの正確な診断は可能なのだろうか?これらの問いに対して,明確な回答は困難であるが,PAHの病態の幅広さが,小児における治療抵抗性PAHの原因の一端を説明しうると,筆者は考えている.
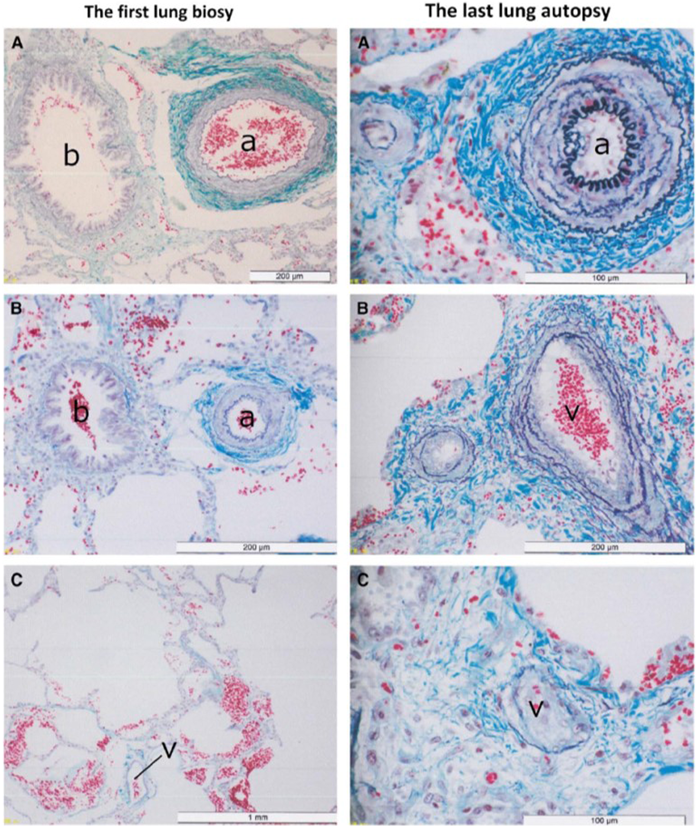
ここで,本邦からの非常に興味深い症例報告を引用する.2017年にMuneuchiらが,急速に進行したPVODにより死亡したTrisomy21の乳児例を報告した57).これによると,動脈管開存症,不完全型心内膜床欠損症を有していたTrisomy21の患児が,動脈管閉鎖術を受けた後,生後4か月時に突然呼吸困難,哺乳不良などをきたし,肺高血圧の増悪と判断された.心臓カテーテル検査では肺静脈閉塞は否定的であった.一酸化窒素(nitric oxide: NO)吸入により肺血流が増加し,肺血管抵抗が低下したため可変PHと考えられた(肺胞出血を合併.明らかな先行感染なし(私信)).その後酸素投与,bosentan, prostaglandin I2(PGI2)持続投与で対応されたものの,肺うっ血,心拡大は進行した.生後2か月時の肺生検所見を根拠に(Fig. 3),PVODではなくPAHの急性増悪と判断され,各種肺血管拡張薬の投与が継続された.しかし,驚くべきことに,生後5か月時に死亡した際の病理解剖では,肺静脈の内膜線維性肥厚および中膜の筋性化,肺小静脈閉塞を認め,PVODと確定診断されたという(Fig. 3).
筆者は最近,Muneuchiらの報告を想起するケースを4症例経験した.2例がTrisomy21(大動脈肺動脈窓・心室中隔欠損症・肺動脈狭窄症合併,完全型心内膜症欠損症・動脈管開存症・II型心房中隔欠損症合併),1例がTrisomy18(心室中隔欠損症・心房中隔欠損症・動脈管開存症・右室二腔症合併),残る1例がVACTERL連合(心室中隔欠損症・部分肺静脈還流異常合併例)であり,3例は心疾患姑息術後(うち1例はマイコプラズマ感染を併発),1例はRSウイルス感染後にPAHの増悪を疑われ,各種肺血管拡張薬を開始,追加あるいは増量したがPHは不変もしくは悪化した.さらに3例で肺胞出血を発症し,かつ全例で肺うっ血,間質性肺疾患を合併した(Fig. 4).うち2例についてはICUでの集中治療を要し,さらにそのうちの1例は喀痰細胞診でへモジデリン貪食マクロファージを認め,肺ヘモジデローシスの合併を確認した.全症例に対してステロイドパルス,その後の後療法を実施した.かつ2例では肺血管拡張薬を減量・中止し,2例では心疾患に対する姑息術・根治術を早期に施行した.これらの対策により全例で呼吸循環状態は徐々に改善し,PH増悪時に急増したKL-6(最高値1,987~6,273 U/mL)も低下傾向となった.
我々が経験したケースでは肺病理像を確認できていないため確定診断が困難であるが,先天性心疾患に伴うPAH(congenital heart disease-PAH: CHD-PAH)が増悪したものと考えて肺血管拡張薬を追加あるいは増量した結果,肺胞出血,肺うっ血をきたし呼吸循環状態が悪化したという経過は,Muneuchiらの報告と共通している.
いったい何が起こっていたのか?これらの症例は,小児重症PAHへの適切な対応を考える上での重要な糸口である.3項および各種の文献報告も踏まえると,小児における重症PAHの一部は純粋なPAHではなく,他の病態とリンクしていることが想定される.4つのポイントに分けて考察する.
①先天異常症候群の合併
Trisomy21の患児は上気道閉塞,睡眠時無呼吸,呼吸器感染症による低酸素血症をきたしやすく,また,不十分な肺胞形成,末梢性肺嚢胞,肺内での気管支動脈–肺動脈吻合などから肺低形成をきたすことが知られている58, 59).また,高度な気腫性変化や線維性変化により肺血管が破壊,閉塞し,肺血管床が減少するとPHを助長する原因となりうる.そして低酸素血症も肺血管攣縮を引き起こし,PHの悪化を助長しうる.かつ,肺ヘモジデローシス症例のうち,20%がTrisomy21であり,さらにその半数以上がPHを合併していたという興味深い報告がある60).PHを合併したTrisomy21症例では,肺毛細血管からの微細な出血が頻繁に生じている可能性がある.
Trisomy21では肺動脈平滑筋細胞の発達が未熟なために,肺動脈圧が上昇した際に中膜が肥厚せずに内膜肥厚が進展し,早期にPAHが進行するのではないかと考えられてきた58).しかし最近,非Trisomy21症例群とTrisomy21症例群で,肺動脈病変の進行スピードに有意差はなかったとの報告が本邦からなされた61).この報告は,Trisomy21におけるPHの診療を行う上で,肺動脈以外の因子にも目を向ける必要があることを示唆している.話が前後するが,6th WSPHにおいても,Trisomy21は3群「PH due to lung disease and/or hypoxia」の小分類,3.5群「Developmental lung disorder」のひとつとされている9).Trisomy21の患児がPHを惹起しうるさまざまな要因を有することを考慮した分類であろう.
Trisomy21と比較すると病態の詳細な検討が不十分であるものの,DiGeorge症候群,VACTREL症候群,CHARGE症候群,Noonan症候群などを合併したCHD患者においても,CHD-PAHが比較的検出されやすいとされている62).また,Trisomy13およびTrisomy18の患児では,肺小動脈の内膜増殖をきたしやすい可能性があることが報告されている63).そして,genetic syndromeを有する患児ではシャント性心疾患による肺血管病変の進行がより急速となりやすいことが指摘されている64).Trisomy21はもちろんのこと,その他のPHが悪化しやすい病態を有する先天異常症候群にも留意する必要がある.
②PAH・PVOD/PCH・間質性肺疾患のoverlap
PVOD/PCHの診断基準には「選択的肺血管拡張薬(エンドセリン受容体拮抗薬,ホスホジエステラーゼ5阻害薬,静注用PGI2)による肺うっ血/肺水腫の誘発」が明記されている65).PVOD/PCHをPAHと診断してしまい,肺血管拡張薬を投与した場合,肺動脈のみを拡張することで換気血流不均衡の助長,肺水腫をきたす可能性がある66).これはPVOD/PCHでしばしば観察される状況であり,PAHとPVOD/PCHの鑑別の難しさを物語っている.また,PVOD/PCHの診断基準では呼吸器疾患に伴う肺高血圧症を除外することとされているが,特発性肺線維症や気腫合併肺線維症においてPVOD/PCH様所見が見られることが複数報告されている67–69).反対に,PVODが肺間質の線維化を惹起することも指摘されている47).PAHとPVOD/PCHのみならず,間質性肺疾患とPVOD/PCHの境界も,これまで考えられていた以上に曖昧である可能性があり,注意を要する.
③PH診療における肺胞出血のリスク
PVODでは肺静脈内膜の細胞性および線維性肥厚,静脈の閉塞・狭窄,筋性動脈化などが特徴である.このような病変からは出血が頻繁に生じ,ヘモジデリン貪食マクロファージが増加することが知られている70).また,前述したように,PAHにおいても肺静脈病変が確認されることが最近報告されているが,そのようなパターンでも出血やヘモジデリン貪食マクロファージが多く見られるとされる71–73).さらに,肺うっ血もびまん性肺胞出血の原因となること,肺胞出血を反復すると肺間質の線維化をきたす可能性があることが示されている74).そして,肺血管拡張薬が肺胞出血を惹き起こす危険性も存在する.少数ではあるが,epoprostenol投与により肺出血をきたしたという報告や75),epoprostenolが抗凝固療法の併用により肺出血のリスクを増加させるとの報告がある76).PGI2が有する強力な血小板凝集抑制作用が関係しているものと推察される77).Sildenafilについても,投与後に肺出血を生じたとの報告が複数存在する78–80).PAH, PVOD/PCHの診療にあたっては肺胞出血,さらにはそれに起因する間質性肺疾患のリスクに注意する必要がある.
④肺血管拡張薬と間質性肺疾患・PVOD/PCH
Epoprostenolが間質性肺炎を惹き起こしたとの報告が複数見られている81–83).特にMorimatsuらの報告では,epoprostenol導入から間質性肺炎が生じるまでにわずか5日間という短期間とのことであった82).また,Volibris®(ambrisentan)の添付文書では重大な副作用として,間質性肺炎の発現もしくは増悪が記載されている.ambrisentanはARTEMIS試験において特発性肺線維症合併PAH症例の病態を悪化させたため,同試験は早期中止となっている84).riociguatについてもRISE-IIP試験において特発性間質性肺炎合併PAHにおいて死亡および呼吸状態の悪化を惹き起こしたため同試験は早期中止とされ85),6th WSPHにおいてもこの2剤はそれぞれの病態において禁忌とされた86).肺血管拡張薬の投与により,なぜ間質性肺疾患が生じるのか,もしくは悪化させるのか,そのメカニズムは明らかではない.アレルギーなどの,薬剤の副作用として間質性肺炎が惹起されたのか,前述のような機序による肺胞出血を介して出現するのか,背景にPVOD/PCHの合併があるのか.症例の蓄積と検討が必要である.
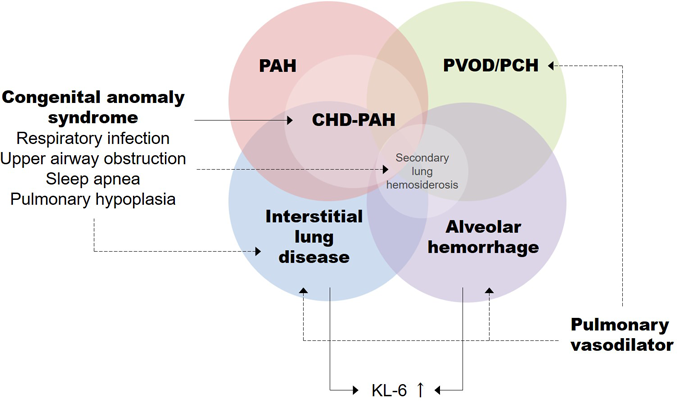
以上をまとめたものがFig. 5となる.すなわち,PAH, PVOD/PCH,間質性肺疾患,肺胞出血は相互に合併しうる病態であり,その発生には先天異常症候群,RSウイルスなどによる呼吸器感染症,上気道狭窄が関与すると考えられる.これらの誘因は特に小児期に見られることが多く,この観点からは,成人発症PAHよりも小児期発症PAHのほうがより多彩な病態を示す可能性があると言える.また,肺血管拡張薬はPVOD/PCH症例もしくは合併例において肺水腫を惹起するのみならず,PVOD/PCHや間質性肺疾患の発生および悪化にも関係している可能性がある.
このメカニズムが全てのPAH, PVOD/PCH,間質性肺疾患に当てはまるわけではない.成人例のPVOD/PCHにおいて肺血管拡張薬が奏功したとの報告や,間質性肺疾患合併PAHに対してsildenafilが有効な可能性があるとの報告もある48, 87, 88).しかし,治療抵抗性の小児PAHに対しては,先天異常症候群を有している可能性,PVOD/PCHや呼吸器疾患によるPHが合併している可能性を考慮し,治療法の選択をより慎重に行う必要があると考えられる.重症例では即座に心臓カテーテル検査を行うわけにもいかず,その鑑別は非常に困難となる.筆者が経験した症例からは,PVOD/PCH,間質性肺疾患,肺胞出血の有無を判断するための胸部高分解能CT(high-resolution computed tomography; HRCT)の撮影やKL-6の測定が,診療方針を決定する上での参考になると考えられる.KL-6は肺胞が傷害されるとII型肺胞上皮細胞で合成が促進されるムコ多糖蛋白であり,間質性肺疾患のバイオマーカーとして知られているが,びまん性肺胞出血においても上昇することが知られている.成人例での検討であるが,KL-6 700 U/mL以上を示したびまん性肺胞出血の症例では生命予後が有意に不良であったとの報告がある89).病態の見極めが困難な重症例においては,肺動脈のみを選択的に拡張させる肺血管拡張薬の経口薬や注射薬の使用を控え,まずはNO吸入療法のみを行うほうがより安全かもしれない.他の肺血管拡張薬とは異なり,NO吸入療法は当然ながら低換気/血流比の部位の血管には届かず,高換気/血流比の部位の血管を拡張させるため,全身の低酸素血症の改善に有利に働く(この点では吸入薬であるiloprostやtreprostinilも有効かもしれない).さらにNO吸入療法については,動物実験において肺動脈のみならず肺静脈の血管抵抗をも低下させたとの報告や90),急性呼吸窮迫症候群の患者において,肺静脈抵抗や肺毛細血管圧を低下させたとの報告があることから91, 92),NO吸入療法はPVOD/PCHに対しても有効である可能性が示唆される.
PAHが重症化し,肺血管拡張薬を投与あるいは増量しても改善しない,むしろ状態が悪化したという状況においては,肺血管拡張薬が不足している可能性だけではなく,肺動脈の向こう側,肺毛細血管や肺静脈病変,肺実質病変の合併の可能性にも思いを巡らせたい.また,PAH症例において肺病理所見を得る機会があった場合には,肺実質病変,肺静脈病変の有無にも注目したいところである.
1項でも述べたように,成人PAH診療においてはcombination therapyが完全に市民権を得た印象がある.しかし本項での検討からは,各種先天異常症候群の存在や,呼吸機能評価が困難であることなどを踏まえると,小児PAHにおけるcombination therapy,特にupfront combination therapyの実施については,慎重に判断すべきであると考える.
本稿の最後に,執筆依頼時に要望いただいた筆者自身の基礎研究について述べることとする.
大学卒後間もない頃にCHD-PAHの患者さん達を受け持ち,そして救命できなかったことを契機に,2009年より東京女子医科大学循環器小児科(現・小児・成人先天性心疾患科)に通いながらPAHの基礎研究に従事させていただくこととなった.
最初の仕事は既知の原因遺伝子が見つかっていないPAH患者において,新規疾患原因遺伝子を同定し,証明することであった.まずは,BMPR2, ACVRL1, SMAD8(SMAD9)等の既知の原因遺伝子が属する,TGF-β/BMPシグナル伝達経路内で候補遺伝子を検索することとなり,PAHの疾患原因遺伝子としてSMAD8を世界ではじめて報告された新谷正樹先生の御指導の下,研究に取り組んだ.2症例で変異が同定されたBMPR1B(ALK6)遺伝子を候補として考え,機能解析を進めたが,western blottingを失敗してばかりでなかなか進められずに苦しんだ.
PAHの原因遺伝子変異の大多数を占めるBMPR2遺伝子変異の場合,BMPシグナリングが低下することで血管平滑筋細胞の増殖を抑制できなくなり,さらに相対的にTGF-βシグナリングが亢進することで血管平滑筋細胞の増殖・成熟が促されると考えられていた20, 93, 94).これを受け,BMPR1B変異もBMPシグナル伝達経路の活性低下をもたらすのであろうと予想したが,どうしてもBMPR1B変異体を導入した細胞からは下流のターゲットであるリン酸化SMAD1/5/8タンパクが強く検出されてしまう.長期出張から帰ってきた新谷先生に「何回やってもリン酸化SMAD1/5/8のバンドが濃く出てしまいます,うまくできません,すみません」とwestern blottingの結果をお見せしたところ,「いや,これは…もしかして(この変異は)gain-of-function(機能獲得型)じゃないの?」と驚かれ,その発想をまるで持っていなかった自分はさらに驚いた.その後のluciferase assayからもwestern blottingでの所見を裏付ける結果が得られた.すなわち,我々が世界で初めて同定したBMPR1B遺伝子変異は明らかにBMPシグナリングを増強させており,前述の定説に反する結果となった95).一方で,BMPR2をノックアウトした肺動脈平滑筋細胞はBMP2・BMP4による刺激でBMPシグナル伝達経路の活性低下を示すものの,BMP6・BMP7による刺激ではその活性増強をもたらすとの報告や96),BMPR2変異を認めないPAH患者の肺動脈平滑筋細胞ではBMPR1Bの発現が有意に増強していたとの報告があり97),これらは我々の研究結果を支持するものであった.
当初は定説通りの結果を出せないことに悩んだが,常識にも多数派にも声の大きい人にも惑わされず,目の前で起こっている事実に冷静に対処し,判断し,表現する大切さを痛感した出来事であった.その後,成人PAH患者においてBMPR1BをターゲットとしているmiR-23b, miR-130aの発現が上昇しており,その発現量が重症度に関与することが報告されるなど98),我々の研究結果を支持する報告が散見されている.
別のPAH2症例で同定したNOTCH3遺伝子変異に関する機能解析を行った時にも多くの壁に阻まれた.野生型あるいは変異型NOTCH3を強制発現させるとその細胞毒性のために数日以内に細胞が死んでしまうため,まずはそれぞれの安定細胞株を樹立しようとさまざまな手法を試したのだが,なかなかうまくできず,細胞塊が全滅することを繰り返した.他の複数の施設にもご相談したが,解決できなかった.その頃たまたま再会した高校同期が分子生物学分野の研究者として大活躍していることを知って相談したところ,非常に具体的な助言をいただき,そのおかげで無事に安定細胞株を樹立し,機能解析を完遂することができた99).
2012年にロサンゼルスで開催された,American Heart Association Scientific Sessionsにはじめて参加した際,当時研究を進める上でのハードルとなっていたヒト肺動脈検体の入手とそれを用いた研究が,いくつかの先進国では容易であることを,ポスター会場でたまたま話しかけてきた初対面のイギリスの基礎研究者に教えてもらった.当時,諸事情で海外留学を諦めつつあったが,やはり自分を試してみたいと奮い立ち,帰国直後から急いで留学調整を始めた.自分がやりたいことができそうだと感じた複数の施設に,メールで履歴書を送ってみたところ「君のBMPR1Bの論文,この前抄読会で読んだよ」とただちにご連絡をくださったケンブリッジ大学呼吸器内科Nicholas Morrell教授の研究室に留学させていただくこととなった.当時の所属先の制約上,1年間という短期間ではあったが,iPS細胞の樹立,血管内皮細胞・平滑筋細胞への分化と機能解析,そして自身が持ち込んだ研究テーマの遂行に全力で取り組んだ.英語力をこき下ろされたり,デスクや研究スペースを他の研究者に奪われたり,空き巣にあったり,不審者に追いかけられたりと嫌なこともたくさんあったが,研究室のメンバーの寛容さに救われ,なんとかやり通すことができた.あの1年間のおかげで,研究者としても医師としても人間としても自分を生まれ変わらせていただいたと今でも確信している.当時の上司や同僚達には論文作成や新規研究の立ち上げについて相談させていただくなど,いまだにお世話になりっぱなしである.なお,この時自身が持ち込んだ研究テーマは,その後の異動による研究中断期間を経て,最近ようやく論文化することができた100).
肺高血圧症の基礎研究に携わるようになってから,断続的にではあるが約10年が経過した.特に研究科学生時代は,基礎研究をしていて楽しいと思う瞬間など1%もなく,ただの苦行の連続であった.それでも,どんなに小さくても新しいことを見つけた時の喜びと興奮は何物にも代えがたいものであった.それが患者さん達のためになるかもしれないとなるとなおさらである.そのふたつは,艱難辛苦に耐え続けるには十分な理由であった.そして,それぞれの研究でそれなりの成果を挙げることができたのは前述した方々,そしてその他にも多くの方々からのサポートをいただいたおかげである.この場をお借りして感謝申し上げたい.本稿を執筆している現在も,偶然居住することになった新たな土地でのさまざまな出会いから大いに刺激をいただき,肺高血圧に関連した研究を立ち上げたところである.臨床現場と基礎研究の狭間にいるからこそ見える景色があるように思う.細々とでも,肺高血圧の患者さん達に直接届くところまで研究を継続していきたいと願っている.
謝辞Acknowledgments
本稿を執筆するにあたり,ご協力を賜りました北海道大学 武田充人先生,福岡市立こども病院 佐川浩一先生,独立行政法人地域医療機能推進機構九州病院 宗内淳先生に心より感謝いたします.
引用文献References
1) Galie N, McLaughlin VV, Rubin LJ, et al: An overview of the 6th World Symposium on Pulmonary Hypertension. Eur Respir J 2019; 53(1): pii:1802148
2) Simonneau G, Montani D, Celermajer DS, et al: Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53(1): pii:1801913
3) Kovacs G, Berghold A, Scheidl S, et al: Pulmonary arterial pressure during rest and exercise in healthy subjects: A systematic review. Eur Respir J 2009; 34: 888–894
4) Heresi GA, Minai OA, Tonelli AR, et al: Clinical characterization and survival of patients with borderline elevation in pulmonary artery pressure. Pulm Circ 2013; 3: 916–925
5) Maron BA, Hess E, Maddox TM, et al: Association of borderline pulmonary hypertension with mortality and hospitalization in a large patient cohort: Insights from the veterans affairs clinical assessment, reporting, and tracking program. Circulation 2016; 133: 1240–1248
6) Rich S, Kaufmann E, Levy PS: The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med 1992; 327: 76–81
7) Sitbon O, Humbert M, Jais X, et al: Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005; 111: 3105–3111
8) Galie N, Channick RN, Frantz RP, et al: Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J 2019; 53(1): pii.1801889
9) Rosenzweig EB, Abman SH, Adatia I, et al: Paediatric pulmonary arterial hypertension: Updates on definition, classification, diagnostics and management. Eur Respir J 2019; 53(1): pii.1801916
10) Douwes JM, Humpl T, Bonnet D, et al: TOPP Investigators: Acute vasodilator response in pediatric pulmonary arterial hypertension: Current clinical practice from the TOPP registry. J Am Coll Cardiol 2016; 67: 1312–1323
11) Nossent EJ, Antigny F, Montani D, et al: Pulmonary vascular remodeling patterns and expression of general control nonderepressible 2 (GCN2) in pulmonary veno-occlusive disease. J Heart Lung Transplant 2018; 37: 647–655
12) Ghigna MR, Guignabert C, Montani D, et al: BMPR2 mutation status influences bronchial vascular changes in pulmonary arterial hypertension. Eur Respir J 2016; 48: 1668–1681
13) Galie N, Humbert M, Vachiery JL, et al: 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 2015; 46: 903–975
14) Douwes JM, Roofthooft MT, Bartelds B, et al: Pulsatile haemodynamic parameters are predictors of survival in paediatric pulmonary arterial hypertension. Int J Cardiol 2013; 168: 1370–1377
15) Loyd JE, Primm RK, Newman JH: Familial primary pulmonary hypertension: Clinical patterns. Am Rev Respir Dis 1984; 129: 194–197
16) Deng Z, Morse JH, Slager SL, et al: Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 2000; 67: 737–744
17) Lane KB, Machado RD, Pauciulo MW, et al: International PPH Consortium: Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet 2000; 26: 81–84
18) Harrison RE, Flanagan JA, Sankelo M, et al: Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet 2003; 40: 865–871
19) Harrison RE, Berger R, Haworth SG, et al: Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation 2005; 111: 435–441
20) Shintani M, Yagi H, Nakayama T, et al: A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J Med Genet 2009; 46: 331–337
21) Trembath RC, Thomson JR, Machado RD, et al: Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345: 325–334
22) Austin ED, Ma L, LeDuc C, et al: Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet 2012; 5: 336–343
23) Ma L, Roman-Campos D, Austin ED, et al: A novel channelopathy in pulmonary arterial hypertension. N Engl J Med 2013; 369: 351–361
24) Olschewski A, Li Y, Tang B, et al: Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ Res 2006; 98: 1072–1080
25) Pardo LA: Voltage-gated potassium channels in cell proliferation. Physiology (Bethesda) 2004; 19: 285–292
26) Simonneau G, Gatzoulis MA, Adatia I, et al: Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62 Suppl: D34–D41
27) Morrell NW, Aldred MA, Chung WK, et al: Genetics and genomics of pulmonary arterial hypertension. Eur Respir J 2019; 53(1): pii.1801899
28) Bongers EM, Duijf PH, van Beersum SE, et al: Mutations in the human TBX4 gene cause small patella syndrome. Am J Hum Genet 2004; 74: 1239–1248
29) Kerstjens-Frederikse WS, Bongers EM, Roofthooft MT, et al: TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J Med Genet 2013; 50: 500–506
30) Zhu N, Gonzaga-Jauregui C, Welch CL, et al: Exome sequencing in children with pulmonary arterial hypertension demonstrates differences compared with adults. Circ Genom Precis Med 2018; 11: e001887
31) Levy M, Eyries M, Szezepanski I, et al: Genetic analyses in a cohort of children with pulmonary hypertension. Eur Respir J 2016; 48: 1118–1126
32) Rosenzweig EB, Morse JH, Knowles JA, et al: Clinical implications of determining BMPR2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J Heart Lung Transplant 2008; 27: 668–674
33) Bohnen MS, Ma L, Zhu N, et al: Loss-of-function ABCC8 mutations in pulmonary arterial hypertension. Circ Genom Precis Med 2018; 11: e002087
34) Richards S, Aziz N, Bale S, et al: ACMG Laboratory Quality Assurance Committee: Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424
35) Machado RD, Aldred MA, James V, et al: Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat 2006; 27: 121–132
36) Cogan JD, Pauciulo MW, Batchman AP, et al: High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med 2006; 174: 590–598
37) Chida A, Shintani M, Yagi H, et al: Outcomes of childhood pulmonary arterial hypertension in BMPR2 and ALK1 mutation carriers. Am J Cardiol 2012; 110: 586–593
38) Girerd B, Montani D, Coulet F, et al: Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med 2010; 181: 851–861
39) Girerd B, Montani D, Eyries M, et al: Absence of influence of gender and BMPR2 mutation type on clinical phenotypes of pulmonary arterial hypertension. Respir Res 2010; 11: 73
40) Sztrymf B, Coulet F, Girerd B, et al: Clinical outcomes of pulmonary arterial hypertension in carriers of BMPR2 mutation. Am J Respir Crit Care Med 2008; 177: 1377–1383
41) Isobe S, Kataoka M, Aimi Y, et al: Improved survival of patients with pulmonary arterial hypertension with BMPR2 mutations in the last decade. Am J Respir Crit Care Med 2016; 193: 1310–1314
42) Evans JD, Girerd B, Montani D, et al: BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir Med 2016; 4: 129–137
43) McLaughlin VV, Archer SL, Badesch DB, et al: ACCF/AHA: ACCF/AHA 2009 expert consensus document on pulmonary hypertension: A report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 2009; 119: 2250–2294
44) Girerd B, Montani D, Jais X, et al: Genetic counselling in a national referral centre for pulmonary hypertension. Eur Respir J 2016; 47: 541–552
45) Hansmann G, Apitz C, Abdul-Khaliq H, et al: Executive summary: Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 2016; 102 Suppl 2: ii86–ii100
46) Abman SH, Hansmann G, Archer SL, et al: American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Surgery and Anesthesia; and the American Thoracic Society: Pediatric Pulmonary Hypertension: Guidelines From the American Heart Association and American Thoracic Society. Circulation 2015; 132: 2037–2099
47) Mandel J, Mark EJ, Hales CA: Pulmonary veno-occlusive disease. Am J Respir Crit Care Med 2000; 162: 1964–1973
48) Montani D, Jais X, Price LC, et al: Cautious epoprostenol therapy is a safe bridge to lung transplantation in pulmonary veno-occlusive disease. Eur Respir J 2009; 34: 1348–1356
49) Wagenvoort CA, Beetstra A, Spijker J: Capillary haemangiomatosis of the lungs. Histopathology 1978; 2: 401–406
50) Eyries M, Montani D, Girerd B, et al: EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet 2014; 46: 65–69
51) Best DH, Sumner KL, Austin ED, et al: EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest 2014; 145: 231–236
52) Pietra GG, Edwards WD, Kay JM, et al: Histopathology of primary pulmonary hypertension: A qualitative and quantitative study of pulmonary blood vessels from 58 patients in the National Heart, Lung, and Blood Institute, Primary Pulmonary Hypertension Registry. Circulation 1989; 80: 1198–1206
53) Hadinnapola C, Bleda M, Haimel M, et al: NIHR BioResource–Rare Diseases Consortium; UK National Cohort Study of Idiopathic and Heritable PAH: Phenotypic characterization of EIF2AK4 mutation carriers in a large cohort of patients diagnosed clinically with pulmonary arterial hypertension. Circulation 2017; 136: 2022–2033
54) Frost A, Badesch D, Gibbs JSR, et al: Diagnosis of pulmonary hypertension. Eur Respir J 2019; 53(1): pii.1801904
55) Overbeek MJ, van Nieuw Amerongen GP, Boonstra A, et al: Possible role of imatinib in clinical pulmonary veno-occlusive disease. Eur Respir J 2008; 32: 232–235
56) Adachi S, Hirashiki A, Kondo T, et al: Imatinib is partially effective for the treatment of pulmonary capillary hemangiomatosis. Intern Med 2014; 53: 603–607
57) Muneuchi J, Oda S, Shimizu D: Rapidly progressive pulmonary veno-occlusive disease in an infant with Down syndrome. Cardiol Young 2017; 27: 1402–1405
58) Saji T: Clinical characteristics of pulmonary arterial hypertension associated with Down syndrome. Pediatr Int 2014; 56: 297–303
59) Bush D, Abman SH, Galambos C: Prominent intrapulmonary bronchopulmonary anastomoses and abnormal lung development in infants and children with Down syndrome. J Pediatr 2017; 180: 156–162.e151
60) Alimi A, Taytard J, Abou Taam R, et al: French RespiRare® group: Pulmonary hemosiderosis in children with Down syndrome: A national experience. Orphanet J Rare Dis 2018; 13: 60
61) Masaki N, Saiki Y, Endo M, et al: Is trisomy 21 a risk factor for rapid progression of pulmonary arteriopathy?: Revisiting histopathological characteristics using 282 lung biopsy specimens. Circ J 2018; 82: 1682–1687
62) Ma L, Chung WK: The genetic basis of pulmonary arterial hypertension. Hum Genet 2014; 133: 471–479
63) Tahara M, Shimozono S, Nitta T, et al: Medial defects of the small pulmonary arteries in fatal pulmonary hypertension in infants with trisomy 13 and trisomy 18. Am J Med Genet A 2014; 164a: 319–323
64) Suzuki K, Yamaki S, Mimori S, et al: Pulmonary vascular disease in Down’s syndrome with complete atrioventricular septal defect. Am J Cardiol 2000; 86: 434–437
65) 日本肺高血圧・肺循環学会「肺静脈閉塞症/肺毛細血管腫症(PVOD/PCH)診療ガイドライン」2017 http://jpcphs.org/publiccomment/guideline2017.pdf
66) Montani D, Lau EM, Dorfmuller P, et al: Pulmonary veno-occlusive disease. Eur Respir J 2016; 47: 1518–1534
67) Sato T, Tsujino I, Tanino M, et al: Broad and heterogeneous vasculopathy in pulmonary fibrosis and emphysema with pulmonary hypertension. Respirol Case Rep 2013; 1: 10–13
68) Awano N, Inomata M, Ikushima S, et al: Histological analysis of vasculopathy associated with pulmonary hypertension in combined pulmonary fibrosis and emphysema: Comparison with idiopathic pulmonary fibrosis or emphysema alone. Histopathology 2017; 70: 896–905
69) Colombat M, Mal H, Groussard O, et al: Pulmonary vascular lesions in end-stage idiopathic pulmonary fibrosis: Histopathologic study on lung explant specimens and correlations with pulmonary hemodynamics. Hum Pathol 2007; 38: 60–65
70) 千葉知宏,植田初江:肺高血圧症の治療抵抗性を病理学的に考察する.最新医学2017; 72: 1109–1116
71) Chazova I, Loyd JE, Zhdanov VS, et al: Pulmonary artery adventitial changes and venous involvement in primary pulmonary hypertension. Am J Pathol 1995; 146: 389–397
72) Jujo T, Sakao S, Ishibashi-Ueda H, et al: Evaluation of the microcirculation in chronic thromboembolic pulmonary hypertension patients: The impact of pulmonary arterial remodeling on postoperative and follow-up pulmonary arterial pressure and vascular resistance. PLoS One 2015; 10: e0133167
73) Dorfmuller P, Humbert M, Perros F, et al: Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum Pathol 2007; 38: 893–902
74) Ioachimescu OC, Stoller JK: Diffuse alveolar hemorrhage: Diagnosing it and finding the cause. Cleve Clin J Med 2008; 75: 264–255
75) 佐地 勉,山田 修,中山智孝,ほか:小児期肺動脈性肺高血圧症症例におけるエポプロステノール治療の有効性と安全性の長期検討—市販後使用成績調査からの検討—.心臓2008; 40: 34–43
76) Ogawa A, Matsubara H, Fujio H, et al: Risk of alveolar hemorrhage in patients with primary pulmonary hypertension: Anticoagulation and epoprostenol therapy. Circ J 2005; 69: 216–220
77) Vane JR, Botting RM: Pharmacodynamic profile of prostacyclin. Am J Cardiol 1995; 75: 3a–10a
78) Yuncu G, Ozkurt S, Sinik Z, et al: Hemoptysis developing soon after use of sildenafil: An observation on two cases. Asian J Androl 2006; 8: 757–758
79) Dixit R, Jakhmola P, Sharma S, et al: Recurrent haemoptysis following sildenafil administration. Indian J Chest Dis Allied Sci 2009; 51: 119–120
80) Pereira e Silva JL, Araujo Neto CA, Marchiori E: Pulmonary hemorrhage after the use of sildenafil. Heart Lung 2012; 41: 407–408
81) Kudelko KT, Nadeau K, Leung AN, et al: Epoprostenol-associated pneumonitis: Diagnostic use of a T-cell proliferation assay. J Heart Lung Transplant 2010; 29: 1071–1075
82) Morimatsu H, Goto K, Matsusaki T, et al: Rapid development of severe interstitial pneumonia caused by epoprostenol in a patient with primary pulmonary hypertension. Anesth Analg 2004; 99: 1205–1207
83) Kesten S, Dainauskas J, McLaughlin V, et al: Development of nonspecific interstitial pneumonitis associated with long-term treatment of primary pulmonary hypertension with prostacyclin. Chest 1999; 116: 566–569
84) Raghu G, Behr J, Brown KK, et al: ARTEMIS-IPF Investigators*: Treatment of idiopathic pulmonary fibrosis with ambrisentan: A parallel, randomized trial. Ann Intern Med 2013; 158: 641–649
85) Nathan SD, Behr J, Cottin V, et al: Idiopathic interstitial pneumonia-associated pulmonary hypertension: A target for therapy? Respir Med 2017; 122 Suppl 1: S10–s13
86) Nathan SD, Barbera JA, Gaine SP, et al: Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J 2018 2019; 53(1): pii.1801914
87) Ogawa A, Miyaji K, Yamadori I, et al: Safety and efficacy of epoprostenol therapy in pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis. Circ J 2012; 76: 1729–1736
88) Han MK, Bach DS, Hagan PG, et al: IPFnet Investigators: Sildenafil preserves exercise capacity in patients with idiopathic pulmonary fibrosis and right-sided ventricular dysfunction. Chest 2013; 143: 1699–1708
89) Kida Y, Ohshimo S, Ota K, et al: KL-6, a Human MUC1 Mucin, as a prognostic marker for diffuse alveolar hemorrhage syndrome. Orphanet J Rare Dis 2012; 7: 99
90) Ayajiki K, Okamura T, Noda K, et al: Functional study on nitroxidergic nerve in isolated dog pulmonary arteries and veins. Jpn J Pharmacol 2002; 89: 197–200
91) Benzing A, Brautigam P, Geiger K, et al: Inhaled nitric oxide reduces pulmonary transvascular albumin flux in patients with acute lung injury. Anesthesiology 1995; 83: 1153–1161
92) Rossetti M, Guenard H, Gabinski C: Effects of nitric oxide inhalation on pulmonary serial vascular resistances in ARDS. Am J Respir Crit Care Med 1996; 154: 1375–1381
93) Newman JH, Phillips JA 3rd, Loyd JE: Narrative review: The enigma of pulmonary arterial hypertension: new insights from genetic studies. Ann Intern Med 2008; 148: 278–283
94) Gu Y, Jin P, Zhang L, et al: Functional analysis of mutations in the kinase domain of the TGF-beta receptor ALK1 reveals different mechanisms for induction of hereditary hemorrhagic telangiectasia. Blood 2006; 107: 1951–1954
95) Chida A, Shintani M, Nakayama T, et al: Missense mutations of the BMPR1B (ALK6) gene in childhood idiopathic pulmonary arterial hypertension. Circ J 2012; 76: 1501–1508
96) Yu PB, Beppu H, Kawai N, et al: Bone morphogenetic protein (BMP) type II receptor deletion reveals BMP ligand-specific gain of signaling in pulmonary artery smooth muscle cells. J Biol Chem 2005; 280: 24443–24450
97) Takeda M, Otsuka F, Nakamura K, et al: Characterization of the bone morphogenetic protein (BMP) system in human pulmonary arterial smooth muscle cells isolated from a sporadic case of primary pulmonary hypertension: roles of BMP type IB receptor (activin receptor-like kinase-6) in the mitotic action. Endocrinology 2004; 145: 4344–4354
98) Wei C, Henderson H, Spradley C, et al: Circulating miRNAs as potential marker for pulmonary hypertension. PLoS One 2013; 8: e64396
99) Chida A, Shintani M, Matsushita Y, et al: Mutations of NOTCH3 in childhood pulmonary arterial hypertension. Mol Genet Genomic Med 2014; 2: 229–239
100) Chida-Nagai A, Shintani M, Sato H, et al: Role of BRCA1-associated protein (BRAP) variant in childhood pulmonary arterial hypertension. PLoS One 2019; 14: e0211450