ここまで知っておきたい心筋症:解剖から分子医学までCardiomyopathies: From Anatomy to Molecular Medicine
富山大学医学部小児科Department of Pediatrics, Toyama University Hospital ◇ Toyama, Japan
発行日:2018年9月1日Published: September 1, 2018
心筋症とは,心筋の器質的あるいは電気的異常を有する多様な疾患群と定義され,原因はしばしば遺伝性である.主な病変が心臓にあるものを一次性心筋症,全身疾患の心筋病変を二次性心筋症と大別される.小児の心筋症では様々な遺伝子に様々な変異があり,遺伝的多様性が特徴である.遺伝形式も常染色体顕性,常染色体潜性,X連鎖性,ミトコンドリア性と様々である.遺伝性心筋症の特徴として,同一遺伝子内の異なる変異は異なる病型を示すこと,遺伝子変異の多くが稀で同一のホットスポットや変異を有することは稀であること,同一の家族内でも様々な浸透率を示すことが挙げられる.また,同一の遺伝子変異を有していたとしても,臨床経過,転帰は同一家族内でも様々である.家族を含めた遺伝学的検査を行うことが重要であり,今後のさらなる心筋症の病態解明が不可欠である.
Cardiomyopathies are defined as abnormalities of the ventricular myocardium. Pediatric cardiomyopathies are rare diseases. Increasingly, the importance of genetic mutations in the pathogenesis of isolated or syndromic pediatric cardiomyopathies is becoming apparent. Pediatric cardiomyopathies are genetically heterogeneous with many different causative genes and multiple mutations in each gene. Variants in the same gene can cause different phenotypes, and variants in different genes can cause the same cardiomyopathy phenotype. There are multiple modes of inheritance for cardiomyopathies, including autosomal dominant, autosomal recessive, X-linked, and mitochondrial. Published guidelines have recommended approaches for genetic testing and family screening in patients with isolated, autosomal-dominant cardiomyopathy. The increasing application of genomic analysis to the pediatric cardiomyopathy population is creating a wealth of information that requires expanded registry participation to further understanding of the pathogenic mechanisms underlying pediatric cardiomyopathies and the genetic, environmental, and other, as of yet undiscovered, modifying factors that impact the severity of disease. This compendium summarizes current knowledge of the genetic and molecular origins of the most common phenotypic presentations of pediatric cardiomyopathies and highlights key areas where additional research is required.
Key words: cardiomyopathy; gene; variant
© 2018 特定非営利活動法人日本小児循環器学会© 2018 Japanese Society of Pediatric Cardiology and Cardiac Surgery
心筋症とは,2006年の米国心臓協会(AHA)では「通常不適切な心室の肥大や拡大を呈するような心筋の器質的あるいは電気的異常を有する多様な疾患群」と定義され,「その原因は多岐にわたるがしばしば遺伝性」である.そして主な病変が心臓にあるものを一次性心筋症,全身疾患の心筋病変を二次性心筋症と大別している(Fig. 1, Table 1).一次性心筋症は遺伝性,混合型(遺伝と後天性),後天性の3つに分類される.
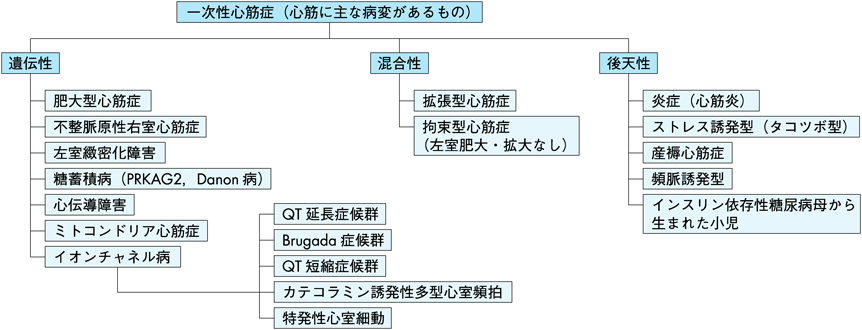
The 2006 American Heart Association classification proposes genetics-based classification; primary cardiomyopathies. Modified from Maron BJ, et al. Circulation 2006; 113: 1807–1816
| 浸潤性 | アミイドーシス,Gaucher病,Hurler病,Hunter病 |
| 蓄積性 | ヘモクロマトーシス,Fabry病,糖原病(Pompe病),Niemann-Pick病 |
| 毒物性 | 薬剤,重金属,化学物質 |
| 心内膜心筋症 | 心内膜線維症,特発性好酸球増多症(Löeffler症候群) |
| 炎症性(肉芽腫) | サルコイドーシス |
| 内分泌性 | 糖尿病,甲状腺機能亢進症,甲状腺機能低下症,副甲状腺機能亢進症,褐色細胞腫,末端肥大症 |
| 頭蓋顔面 | Noonan症候群,多発性黒子症候群 |
| 神経・筋疾患 | Friedreich失調症,Duchenne–Becker筋ジストロフィー,Emery–Dreifuss筋ジストロフィー,筋強直性ジストロフィー,神経線維腫症,結節性硬化症 |
| 栄養障害 | 脚気衝心,ペラグラ,壊血病,セレン,カルニチン,Kwashiorkor |
| 自己免疫・膠原病 | 全身エリテマトーデス,皮膚筋炎,関節リウマチ,強皮症,結節性多発動脈炎 |
| 電解質異常 | |
| 癌治療後 | アントラサイクリン系,シクロフォスファミド,放射線 |
| The 2006 American Heart Association classification proposes genetics-based classification; secondary cardiomyopathies. Modified from Maron BJ, et al. Circulation 2006; 113: 1807–1816 | |
本稿では,心筋の微細構造について概説し,一次性心筋症として,肥大型拡張症,拡張型心筋症,心筋緻密化障害,ミトコンドリア心筋症と二次性心筋症の遺伝学的要因について概説する.
心臓には主に心筋細胞,線維芽細胞,血管内皮細胞の3種類の細胞が存在する.心筋細胞が心臓体積のほぼ90%を占めるが,細胞数ではこれらの3種はほぼ同等とも考えられている.心筋細胞の周りには線維芽細胞が産生したコラーゲンやラミニンなどの結合組織が豊富に存在し,さらに毛細血管が心筋細胞に酸素や栄養素を供給している.
心筋細胞は細胞質に豊富な筋原線維(myofibril)をもち,休むことなく収縮-弛緩を繰り返すことに分化した細胞である.心筋細胞には基本的な収縮ユニットである筋原線維と,興奮収縮連関に重要なカルシウムイオンの貯蔵庫である筋小胞体(sarcoplasmic reticulum),心筋収縮に必要なATPの供給源であるミトコンドリア,核(多核)が存在する(Fig. 2).
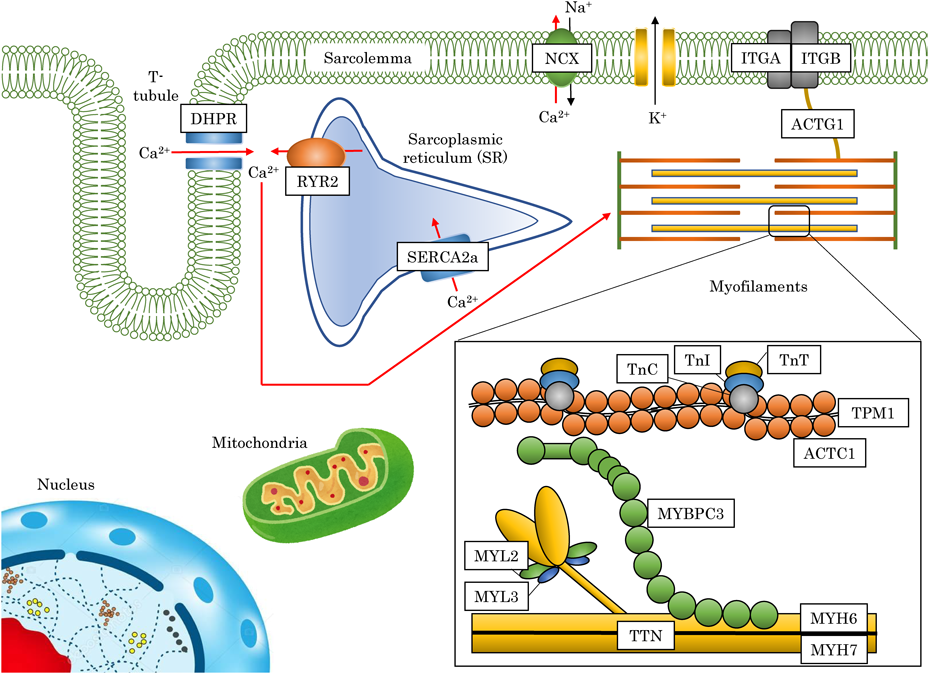
心筋細胞は,ミオシンを主成分とするthick filamentとアクチンを主成分とするthin filament,が高度に組織化された筋原線維により満たされている.アクチン線維が付着する2本のZ帯に挟まれた2.2 µm(静止時)の収縮単位は,サルコメア(sarcomere)と呼ばれる.
Ca2+非存在下ではトロポニン複合体は密着し合い,トロポニンIはアクチンと結合してアクチン-ミオシン結合部位がトロポミオシンによりブロックされる.Ca2+存在下では,Ca2+が結合したトロポニンCに構造変化が起こり,トロポニンI–アクチン間の結合を解離し,トロポミオシンはアクチンの溝から移動してアクチン-ミオシン結合部位が解放されるようになり,収縮が開始する.
アクチンやミオシンはα-アクチニン(α-actinin)とデスミン(desmin)を主成分とするZ帯に強固に固定されている.さらにZ帯は細胞膜に接する部分でコスタメア(costamere)と呼ばれる特殊な構造(接着斑)を形成する.コスタメアにはビンキュリン(vinculin),ターリン(talin)などの細胞骨格蛋白とともに,focal adhesion kinase(FAK)やmuscle LIM proteinなどのシグナル伝達蛋白質が存在する.Z帯は細胞-基質接着因子であるインテグリン(integrin)を介して,細胞外のコラーゲン,フィブロネクチン,ラミニンなどの基質(extracellular matrix)と接着し,筋原線維の収縮を隣り合う細胞に伝えるとともに,細胞外の機械的なストレスを核に伝えるシグナル伝達の場にもなっている.また心筋細胞の両端においてはZ帯は介在板(intercalated disk)を形成し,細胞-細胞接着因子であるカドヘリン(cadherin)やカテニン(catenin)を介して隣り合う心筋細胞同士を強固に接着している.
肥大型心筋症は,主に心室中隔を中心とした心室壁の肥厚により,拡張能の低下をきたす疾患である.心室の収縮性はむしろ過度に亢進している場合が多く,左室流出路閉塞が見られることがある.無症状に経過する場合も多く,罹患率は一般人口の0.2%と高頻度の報告がある1).小児期から若年成人期発症例では,乳児期に1つのピークがあり,それ以降は10歳から25歳で発症するものが多い.乳児期の肥大型心筋症は,Noonan症候群など全身疾患や症候群に合併するものが多く,小児期以降の発症例は家族性の肥大型心筋症が多い.肥大型心筋症は若年者の突然死の原因として最も重要な疾患である.早期には拡張不全による心不全をきたすが,経過とともに左室の拡張と収縮性の低下をきたす場合がある.組織学的には,心筋細胞の不均一な肥大と錯綜配列を特徴としている.心室中隔の肥大が著明な例が多く(非対称性中隔肥大),著しい症例では左室流出路狭窄をきたす(閉塞性肥大型心筋症).心筋肥大により,心室内腔は狭小化し,拡張障害を示す.また,心拍出量の低下は,失神や突然死をきたすことがあり,心筋虚血のため,胸痛を生じることもある.収縮力は,正常に保たれている例が多いが,次第に心拡大と収縮力の低下をきたし心不全を生じる例(拡張相肥大型心筋症)は,予後不良である.
肥大型心筋症は主にサルコメアの疾患であり,心筋の収縮や調節を行う8つのサルコメア関連遺伝子の異常がコホート研究の60%において認められる(Table 2).細胞レベルでは,肥大型心筋症の変異はマイオフィラメントの感受性の増加やカルシウムの結合性の増加,actin-activated ATPaseの活性の増加が見られる.動力の発生において,肥大型心筋症の遺伝子変異はエネルギー非効率化とエネルギー消費の増大をもたらす.拡張型心筋症や不整脈源性右室異形成症と同じように,遺伝形式は常染色体顕性遺伝であり,座と対立遺伝子の異質性(locus and allelic heterogeneity)が認められる.肥大型心筋症に最も変異が見られる遺伝子はMYH7とMYBPC3であり,2つで50%を占める.残りのサルコメア遺伝子のTNNT2, TNNI3, TPM1, ACTC1, MYL3, MYL2で10%を占める.近年,複合ヘテロ接合性や多因子遺伝病が肥大型心筋症で2.5~5%の割合で見いだされるようになってきている.このような患者は若年発症でより重症であり,心筋切除術やICD埋め込み術の割合が高い.病原遺伝子に加えて,遺伝子的要素,エピジェネティックな因子,環境因子の修飾が肥大型心筋症の病型には重要である.
| Gene | Protein | Locus | Inheritance | Prevalence (%) |
|---|---|---|---|---|
| Sarcomere | ||||
| MYH7 | β-Myosin heavy chain | 14q11.2-q12 | AD | 25–40 |
| MYH6 | α-Myosin heavy chain | 14q11.2-q12 | AD | <1 |
| MYL2 | Regulatory myosin light chain | 12q23-q24.3 | AD | <1 |
| MYL3 | Essential myosin light chain | 3p21.2-p21.3 | AD | <1 |
| TNNT2 | Cardiac troponin T | 1q32 | AD | 3–5 |
| TNNI3 | Cardiac troponin I | 19p13.4 | AD | 1–5 |
| TPM1 | α-Tropomyosin | 15q22.1 | AD | 1–5 |
| ACTC | α-Cardiac actin | 15q14 | AD | <1 |
| TNNC1 | Cardiac troponin C | 3p21.1 | AD | <1 |
| MYBPC3 | Cardiac myosin-binding protein C | 11p11.2 | AD | 25–40 |
| TTN | Titin | 2q31 | AD | <1 |
| Z-disc | ||||
| ACTN2 | α-Actinin 2 | 1q42-q43 | AD | <1 |
| ANKRD1 | Cardiac ankyrin repeat protein | 10q23.31 | AD | <1 |
| CSPR3 | Muscle LIM protein | 11p15.1 | AD | <1 |
| LDB3 | LIM binding domain 3 | 10q22.2-q23.3 | AD | 1–5 |
| MYOZ2 | Myozenin 2 | 4q26-q27 | AD | <1 |
| TCAP | Telethonin | 17q12-q21.1 | AD | <1 |
| VCL | Vinculin/metavinculin | 10q22.1-q23 | AD | <1 |
| Calcium-handling | ||||
| JPH2 | Junctophilin 2 | 20q13.12 | AD | <1 |
| PLN | Phospholamban | 6q22.1 | AD | <1 |
MYBPC3の遺伝子変異は,肥大型心筋症では最もよく認められる遺伝子変異である.MYBPC3は,ミオシンを介した収縮の安定化や調節を行っていると考えられている.ほかの肥大型心筋症の遺伝子変異ではミスセンス変異によりアミノ酸が置換されるのとは対照的に,MYBPC3の多くはナンセンス変異かフレームシフトであり,不完全な蛋白が産生される2).MYBPC3の遺伝子変異は浸透率が低く,発症が晩発性であり,生存率は良いが,いったん発症するとリスクは他の肥大型心筋症と同等である3, 4).
MYH7の遺伝子変異は肥大型心筋症全体の原因の20%を占める.一般的に,MYH7遺伝子変異は,著明な肥大を伴う,比較的若年に発症する典型的な肥大型心筋症と関連がある5).MYH7の遺伝子変異は肥大型心筋症だけに限らず,拡張型心筋症や心筋緻密化障害,ミオパチーにも見られる.
thin filamentのトロポニン複合蛋白をコードするTNNT2とTNNI3遺伝子にも病型の多様性があり,肥大型心筋症,拡張型心筋症,拘束型心筋症,心筋緻密化障害にも認められる6).他の多くのサルコメア蛋白のように,TNNT2変異はしばしば比較的軽症の拡張型心筋症を示すが心臓突然死のリスクは高い.トロポニンの遺伝子変異によりthin filament上のカルシウム結合能結合能は肥大型心筋症の場合は増強し,拡張型心筋症の場合は減弱する7).
肥大型心筋症の他の4つのTPM1, ACTC1, MYL2, MYL3遺伝子変異は良性である.TPM1の遺伝子変異は拡張型心筋症や肥大型心筋症を引き起こす8, 9).肥大型心筋症の遺伝子変異はほとんどがN末端のドメインやトロポニンT結合部位に集積しthin filamentのカルシウム感受性を増加させる.
拡張型心筋症は,左室の拡張と収縮性の低下が特徴である.心筋の変性,萎縮や線維化により,心収縮力が低下し,心拍出量が低下する.小児期では,いずれの年齢にも認められる.原因疾患にかかわらず,5年生存率は50%と不良である10, 11).家族性,心筋炎後など原因は様々である.交感神経系の亢進により,後負荷は増大し,心筋酸素消費量が増大する.TNF-αなど種々のサイトカインが血中に増加し,心筋障害を助長する.死亡原因は,ポンプ機能の低下による心不全か,不整脈による突然死である.近年は,ACE阻害薬やβ遮断薬など内科的治療に加えて,植込み型除細動器や心室補助装置の普及により予後はいくぶん改善されている.
拡張型心筋症の約30~40%に家族性があり,小児期ではミトコンドリア遺伝や常染色体顕性遺伝形式をとるものが主体であり,X連鎖性や常染色体潜性遺伝形式はまれである.
家族性拡張型心筋症については,50以上の原因遺伝子が示唆されている(Table 3).家族性拡張型心筋症の30~35%に遺伝子変異が見いだされており,TTN, LMNA, MYH7, TNNT2が主要な遺伝子である.TTNの遺伝子変異が最もよく見いだされており,家族性拡張型心筋症の25%,弧発性拡張型心筋症の18%で報告されている12).伝導障害を伴う拡張型心筋症の場合,LMNA遺伝子変異が1/3に認められる12).
| Gene | Protein | Locus | Inheritance | Prevalnece | Phenotype |
|---|---|---|---|---|---|
| Sarcomere | |||||
| MYH7 | Myosin heavy chain 7 | 14q12 | AD | 4% | HCM/DCM/RCM/LVNC |
| MYH6 | Myosin heavy chain 6 | 14q12 | AD | HCM/DCM | |
| MYBPC3 | Myosin binding protein C, cardiac | 11p11.2 | AD | 2% | HCM/DCM/LVNC |
| TNNT2 | Troponin T2, cardiac type | 1q32 | AD | 2% | HCM/DCM/RCM/LVNC |
| TNNC1 | Troponin C1, slow skeletal and cardiac type | 3p21.1 | AD | HCM/DCM | |
| TNNI3 | Troponin I3, cardiac type | 19q13.4 | AD, AR | HCM/DCM/RCM | |
| TPM1 | Tropomyosin 1 (alpha) | 15q22.1 | AD | HCM/DCM/RCM/LVNC | |
| ACTC1 | Actin, alpha, cardiac muscle 1 | 15q14 | AD | HCM/DCM/LVNC | |
| TTN | Titin | 2q31 | AD | 20–25% | HCM/DCM/ARVC |
| Z-disc | |||||
| BAG3 | BCL2 associated athanogene 3 | 10q25.2-q26.2 | AD | 2% | DCM |
| CRYAB | Crystallin alpha B | 11q23.1 | AD | DCM/HCM | |
| FHL2 | FHL2 four and a half LIM domains 2 | 2q12.2 | AD | ||
| MURC | Muscle related coiled-coil protein | 9q31.1 | AD | DCM | |
| MYPN | Myopalladin | 10q21.3 | AD | 2% | DCM/HCM/RCM |
| NEBL | Nebulette | 10p12 | AD | DCM/EFE | |
| NEXN | Nexilin F-actin binding protein | 1p31.1 | AD | DCM/HCM | |
| Cell structure | |||||
| ACTN2 | Actinin alpha 2 | 1q42-q43 | AD | DCM | |
| CSRP3 | Cysteine and glycine rich protein 3 | 11p15.1 | AD | HCM/DCM | |
| DMD | Dystrophin | Xp21.2 | XR | DCM | |
| FKTN | Fukutin | 9q31.2 | AR | DCM | |
| JUP | Junction plakoglobin | 17q21 | AR, AD | DCM/ARVC | |
| LDB3 | LIM domain binding 3 | 10q22.3-q23.2 | AD | DCM/LVNC | |
| SGCD | Sarcoglycan delta | 5q33.3 | AD | DCM | |
| TCAP | Titin-cap | 17q12 | AD | HCM/DCM | |
| VCL | Vinculin | 10q22-q23 | AD | HCM/DCM | |
| Desmosome | |||||
| DES | Desmin | 2q35 | AD | DCM/RCM | |
| DSP | Desmoplakin | 6p24 | AR | DCM/ARVC/LVNC | |
| PLN | Phospholamban | 6q22.1 | AD | DCM | |
| EMD | Emerin | Xq28 | XR | DCM | |
| LMNA | Lamin A/C | 1q22 | AD | 6% | DCM |
| TMPO | Thymopoietin | 12q22 | AD | DCM | |
| ANKRD1 | Ankyrin repeat domain 1 | 10q23.31 | AD | HCM/DCM | |
| GATAD1 | GATA zinc finger domain containing 1 | 7q21-q22 | AR | DCM | |
| RBM20 | RNA binding motif protein 20 | 10q25.2 | AD | 2% | DCM |
| Others | |||||
| ABCC9 | ATP binding cassette subfamily C member 9 | 12p12.1 | AD | 1% | DCM |
| SCN5A | Sodium voltage-gated channel alpha subunit 5 | 3p21 | AD | 2% | DCM |
| LAMA4 | Laminin subunit alpha 4 | 6q21 | AD | DCM | |
| DOLK | Dolichol kinase | 9q34.11 | AR | DCM | |
伝導障害を伴う拡張型心筋症では,lamin A/C(LMNA)遺伝子変異が認められる13).LMNA変異により引き起こされる拡張型心筋症は,初期には房室ブロックなどの伝導障害が認められること,心臓突然死のリスクが高いことが特徴的である14).一般的に伝導障害は拡張型心筋症の病勢に伴い進行するが,骨格筋の障害との関連は症例ごとに異なる13).
骨格筋異常を伴う拡張型心筋症は,ジストロフィンの遺伝子変異が原因であることが多い15).ジストロフィン遺伝子はDuchenneやBecker muscular dystrophy(DMD/BMD)の責任遺伝子でもある.DMDやBMDは骨格筋疾患の多くを占め,いずれも小児期あるいは若年成人で発症するが,ほとんどの症例は25歳までに拡張型心筋症を発症する.ジストロフィンは細胞骨格蛋白であり,格子状のネットワークを形成して,筋細胞を支持している.そして,サルコメアと筋細胞膜や細胞外基質との結合と収縮力の伝達において,最も重要な役割を果たしている.さらに,NO合成酵素と作用して,細胞シグナル伝達にも関与している16).ジストロフィンのN末端はサルコメアのアクチンと結合し,C末端はジストログリカン,サルコグリカン,シントロピン,ジストロブレビンなどのジストロフィン関連蛋白と結合している.さらに,これらの蛋白は,α-ジストログリカンを通じて,ラミニン(laminin)や細胞外基質と結びついている.これらジストロフィン関連蛋白の異常は,ジストロフィン同様に骨格筋疾患や心筋症の原因となる.
左室心筋緻密化障害は,心室壁の過剰な網目状の肉柱形成と深い間隙を形態的特徴とし,WHO分類ではunclassified cardiomyopathyの1つとして分類されている3).典型例は,新生児期に心不全のため死亡し,心移植の対象になっている疾患である17).血行動態の特徴は心収縮力の低下で,左室の拡大を伴う例と,伴わないものがある.拘束型心筋症に類似した血行動態を呈する症例も報告されている.収縮力の低下している網目状の肉柱の間に,血栓が形成されやすく,他の拡張型心筋症に比し,脳や肺などの,全身の血栓塞栓症を合併する危険性が高い.臨床像は無症状の症例から,高度の心機能障害を有し,心移植の対象になっているものまであり,きわめて多彩である17–19).原因は多岐にわたる.
弧発性と家族性の双方認められる.本邦の全国調査の結果では,高率(40%)に家族例が認められ,X連鎖性のほか,常染色体顕性遺伝形式,常染色体潜性遺伝形式,あるいはミトコンドリア遺伝子変異が疑われる家系もあり,遺伝的多様性が特徴である18).TAZ, DTNAやLDB3などの多数の遺伝子変異が原因であることがわかっている(Table 4).
| Gene | Protein | Locus | Inheritance |
|---|---|---|---|
| Genes associated with cardiomyopathies and or CHD and LVNC | |||
| MYH7 | Beta-myosin heavy chain 7 | 14q12 | AD |
| MYBPC3 | Myosin-binding protein C | 11p11.2 | AD |
| TNNT2 | Cardiac troponin T2 | 1q32 | AD |
| TPM1 | Tropomyosin 1 | 15q22.1 | AD |
| ACTC1 | Cardiac alpha-actin | 15q14 | AD |
| ACTN2 | Alpha-actinin 2 | 1q42-q43 | AD |
| DTNA | Dystrobrevin | 18q12.1 | AD |
| LDB3 | Z-Band alternatively spliced PDZ motif-containing protein | 10q22.3-q23.2 | AD |
| DMPK | Dystrophia myotonica protein kinase | 19q13.32 | AD |
| DSP | Desmoplakin | 6p24 | AD |
| PKP2 | Plakophilin 2 | 12p11.21 | AR |
| RYR2 | RYR2 | 1q43 | AD |
| LMNA | Lamin AC | 1q22 | AD |
| MIB1 | Mindbomb, homolog of, Drosophila | 18q11.2 | AD |
| PRDM16 | PR domain protein 16 | 1p36.32 | AD |
| TBX20 | T-box20 | 7p14.2 | AD |
| ABCC9 | ATP-binding cassette, subfamily C members | 12p12.1 | AD |
| CASQ2 | Calsequestrin 2 | 1p13.1 | AR |
| HCN4 | Hyperpolarization-activated cyclic nucleotide-gated potassium channel 4 | 15q24.1 | AD |
| SCN5A | Sodium channel, voltage-gated, type V, alpha subunit | 3p21 | AD |
| PLEKHM2 | Pleckstrin homology domain-containing protein, family M, member 2 | 1p36.21 | AD |
| Monogenic syndromes with major extracardiac traits and cardiac involvement including LVNC | |||
| ARFGEF2 | ADP-ribosylation factor guanine nucleotide-exchange factor 2 | 20q13.13 | AR |
| MLYCD | Malonyl–coenzyme A decarboxylase deficiency | 16q23.3 | AR |
| MMACHC | Cobalamin C deficiency | 1p34.1 | AR |
| NNT | Nicotinamide nucleotide transhydrogenase | 5p12 | AR |
| NSD1 | Nuclear receptor binding SET domain protein1 | 5q35.3 | Isolated type |
| RSK2 | Ribosomal S6 kinase 2 | Xp22.12 | XLD |
| YWHAE (14-3-3ε) | Monooxygenase activation protein, epsilon isoform | 17p13.3 | AD |
小児と成人の心筋緻密化障害では,原因遺伝子にも差が見られ,成人では,サルコメア遺伝子変異が主体で,TAZ, LDB3などの小児で見られる遺伝子変異は,まれである.単一遺伝子変異のほかにも,染色体異常,筋疾患,ミトコンドリア筋症などの疾患に合併して見られることがあり,原因は多岐にわたる25–30).近年,スペイン人の心筋緻密化障害の2家系から,常染色体顕性遺伝形式でMIB1の遺伝子変異が認められた.このMIB1のノックアウトマウスでは,心筋に著明な肉柱化と緻密化障害が認められ,心筋緻密化障害とNOTCH1シグナルとの関連が示唆されている31).
ミトコンドリア病は心筋症を合併する.ミトコンドリア病の症状は様々で,通常は多臓器に障害が見られ,特に高容量のATPを必要とする心臓,骨格筋,神経が侵される.典型的にはその変化は乳児や胎児期に起こり,サルコメア遺伝子変異の心筋症に見られるような代償期はない.ミトコンドリア心筋症は,肥大型心筋症様の病型がより一般的である.いくつかの全身性のミトコンドリア病(ミトコンドリア脳筋症,MELAS, MERRF, Kearns-Sayr症候群,Leigh病など)では,ミオパチー,脳症,てんかん,眼筋麻痺,失調症,難聴といったミトコンドリア病の症状とともに,その特徴に含まれる.
ミトコンドリアの機能異常はミトコンドリアや核の遺伝子変異により引き起こされる(Fig. 3).ミトコンドリアDNAの異常はheteroplasmy(1細胞中における変異ミトコンドリアDNAの混在)が特徴とされる.遺伝形式は母系である.核の遺伝子変異に由来するミトコンドリア病は様々な遺伝子形式を示し,典型的には常染色体潜性遺伝やX染色体遺伝を示すことが多い.
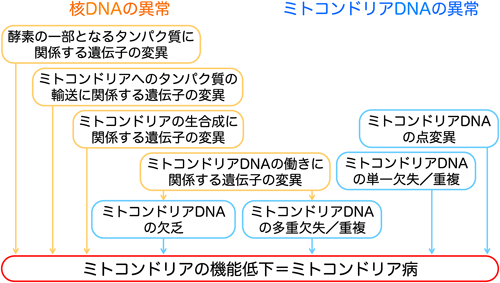
ミトコンドリア病の心筋症の頻度は17~40%と様々で,多くは肥大型心筋症である32–34).核蛋白をコードするSCO2の遺伝子変異は,乳児のミトコンドリア心筋症の原因としてよく知られている35).SCO2はシトクロームC酸化酵素,呼吸鎖の複合体IVの一部であり,新生児期に肥大型心筋症や脳症や筋緊張低下を発症する36).ほかには,ミトコンドリア心筋症を引き起こす核蛋白遺伝子は,ミトコンドリアの呼吸鎖を構成する遺伝子(NDUFS2, NDUFV2, SDHA, SCO2, COX15, TMEM70等),ミトコンドリアDNAの翻訳に関連する遺伝子(MRPS22),ATP合成に関連する遺伝子(SLC25A3),リン脂質のリモデリングに関連する遺伝子(TAZ)がある.TMEM70はミトコンドリア複合体Vの欠損からATP合成を減少させ,筋緊張低下,乳酸アシドーシス,3-methylaciduriaを合併し37),心筋症としては,肥大型心筋症,X連鎖性心内膜線維弾性症,X連鎖性致死的拡張型心筋症,家族性の心筋緻密化障害を引き起こす.心室性不整脈が特徴的である.
Barth症候群は,X連鎖性の心筋骨格筋疾患であり,成長障害好中球減少,3 -メチルグルタコンサン尿が認められる.電子顕微鏡では,筋細胞のミトコンドリアの機能異常が認められる.一般的には,乳児早期に発症し,心不全や敗血症で死亡するが,乳児期以降では,拡張型心筋症は存在するが比較的安定した臨床経過であることが多い.Barth症候群の責任遺伝子はTAZ遺伝子であり,この遺伝子変異のためミトコンドリア膜上のカルジオリピンの欠乏が引き起こされる.TAZ遺伝子変異は,拡張型心筋症,心内膜線維弾性症や心筋緻密化障害など,多彩な心筋症を発症するが,Barth症候群の臨床症状を伴う場合も伴わない場合もある38).
ミトコンドリアゲノムは16.6 kbであり,そのなかで,全身性の疾患の原因とされる250以上のミトコンドリアDNAの遺伝子変異が報告されている39).MTTL1のm.3243 A>Gの変異はMELAS患者に認められ,脳症,低身長,梗塞様のエピソード,けいれん,乳酸アシドーシス,半盲を合併する.MELAS患者の心筋症は40%に認められ,心機能低下を伴った対称性の肥大をきたす40).MERRFの患者は80~90%の割合で肥大型心筋症もしくは拡張型心筋症を合併する41).MTTIの変異は肥大型心筋症および拡張型心筋症を発症する42).MTTGの変異は,肥大型心筋症を発症する43).
種々の代謝疾患は,乳幼児発症の心筋症を合併する.代謝疾患は心臓への蓄積もしくは浸潤する疾患,エネルギー代謝異常症,心毒性のある代謝産物を産生する疾患に分類される44).
Pompe病は,グリコーゲンを分解するリソゾーム酵素であるGAAの遺伝子変異による,acidα-glucosidaseの欠損症である.肥大型心筋症とミオパチーを主症状とする.乳児型および遅発型(小児型・成人型)に分類され,乳児型は肥大型心筋症により,遅発型は呼吸不全により生命が脅かされる.Pompe病に対し酵素補充療法が導入され,心筋肥厚が改善するため,乳児型の生命予後が改善されてきている.
Fabry病の原因であるGLAの遺伝子変異は,全身性のX連鎖性のライソゾームの蓄積障害を引き起こし,心筋症,不整脈,腎障害,肢端触覚異常や被角血管腫,毛細血管拡張症といった皮膚症状,発汗低下,角膜混濁や脳血管障害を特徴とする.Fabry病はライソゾームのhydrolase α-galactosidase Aの欠損や欠乏により,glycosphingolipidが蓄積することが原因である.典型的には男性に認められるが,まれに女性患者にも認められる.GLA変異は肥大型心筋症を有した家系の3%に認められた45).Fabry病は,心筋症が唯一の症状であり,酵素補充療法により治療が可能である.
PRKAG2の遺伝子変異は,グリコーゲンの蓄積や心筋細胞の錯綜配列により,常染色体顕性遺伝の形式で,家族性の肥大型心筋症を引き起こす46).PRKAG2の遺伝子変異は,心電図上,心室の早期興奮,徐脈傾向,進行性の伝導障害を伴ったWPW症候群も合併する.
Danon病は,X連鎖性の肥大型心筋症であり,ミオパチー,知的障害を伴う.LAMP2の遺伝子変異により,ライソゾームの膜レセプターを介したオートファジーが原因とされている.Danon病は,心室の早期興奮,不整脈,拡張型心筋症へと進行する著明な心筋肥大を特徴とし,通常の肥大型心筋症より予後不良であり,成人期早期にしばしば心移植が必要となる.Danon病は女性においてより重症である.
次世代シーケンサーの登場により,遺伝学的要因が徐々に明らかにされてきているが,遺伝子・表現型相関が明らかであるのはごく一部にすぎない.また,心筋症の診療において,遺伝子診断が行われているのは少数の症例に限られる.また,今後の心筋症研究の発展のために,また,診療において遺伝性心筋症の原因探索のために,大きな家系の集積および正確な臨床情報収集が不可欠である.
本論文について,開示すべき利益相反(COI)はない.
1) Maron BJ: Hypertrophic cardiomyopathy. Lancet 1997; 350: 127–133
2) Marston S, Copeland O, Jacques A, et al: Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res 2009; 105: 219–222
3) Niimura H, Bachinski LL, Sangwatanaroj S, et al: Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med 1998; 338: 1248–1257
4) Charron P, Dubourg O, Desnos M, et al: Clinical features and prognostic implications of familial hypertrophic cardiomyopathy related to the cardiac myosin-binding protein C gene. Circulation 1998; 97: 2230–2236
5) Watkins H, Rosenzweig A, Hwang DS, et al: Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med 1992; 326: 1108–1114
6) Kimura A, Harada H, Park JE, et al: Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet 1997; 16: 379–382
7) Robinson P, Griffiths PJ, Watkins H, et al: Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res 2007; 101: 1266–1273
8) Thierfelder L, Watkins H, MacRae C, et al: Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: A disease of the sarcomere. Cell 1994; 77: 701–712
9) Watkins H, McKenna WJ, Thierfelder L, et al: Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med 1995; 332: 1058–1064
10) Matitiau A, Perez-Atayde A, Sanders SP, et al: Infantile dilated cardiomyopathy. Relation of outcome to left ventricular mechanics, hemodynamics, and histology at the time of presentation. Circulation 1994; 90: 1310–1318
11) Lipshultz SE, Sleeper LA, Towbin JA, et al: The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med 2003; 348: 1647–1655
12) Herman DS, Lam L, Taylor MR, et al: Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012; 366: 619–628
13) Brodsky GL, Muntoni F, Miocic S, et al: Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation 2000; 101: 473–476
14) Fatkin D, Roy P, Morgan JJ, et al: Percutaneous balloon mitral valvotomy with the Inoue single-balloon catheter: Commissural morphology as a determinant of outcome. J Am Coll Cardiol 1993; 21: 390–397
15) Hoffman EP, Brown RH Jr., Kunkel LM: Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 1987; 51: 919–928
16) Chang WJ, Iannaccone ST, Lau KS, et al: Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc Natl Acad Sci USA 1996; 93: 9142–9147
17) Chin TK, Perloff JK, Williams RG, et al: Isolated noncompaction of left ventricular myocardium: A study of eight cases. Circulation 1990; 82: 507–513
18) Ichida F, Hamamichi Y, Miyawaki T, et al: Clinical features of isolated noncompaction of the ventricular myocardium: Long-term clinical course, hemodynamic properties, and genetic background. J Am Coll Cardiol 1999; 34: 233–240
19) Pignatelli RH, McMahon CJ, Dreyer WJ, et al: Clinical characterization of left ventricular noncompaction in children: A relatively common form of cardiomyopathy. Circulation 2003; 108: 2672–2678
20) Hoedemaekers YM, Caliskan K, Michels M, et al: The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ Cardiovasc Genet 2010; 3: 232–239
21) Klaassen S, Probst S, Oechslin E, et al: Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation 2008; 117: 2893–2901
22) Dellefave LM, Pytel P, Mewborn S, et al: Sarcomere mutations in cardiomyopathy with left ventricular hypertrabeculation. Circ Cardiovasc Genet 2009; 2: 442–449
23) Budde BS, Binner P, Waldmuller S, et al: Noncompaction of the ventricular myocardium is associated with a de novo mutation in the beta-myosin heavy chain gene. PLoS One 2007; 2: e1362
24) Kelley-Hedgepeth A, Towbin JA, Maron MS: Images in cardiovascular medicine. Overlapping phenotypes: Left ventricular noncompaction and hypertrophic cardiomyopathy. Circulation 2009; 119: e588–e589
25) Wong JA, Bofinger MK: Noncompaction of the ventricular myocardium in Melnick-Needles syndrome. Am J Med Genet 1997; 71: 72–75
26) Mandel K, Grunebaum E, Benson L: Noncompaction of the myocardium associated with Roifman syndrome. Cardiol Young 2001; 11: 240–243
27) Corrado G, Checcarelli N, Santarone M, et al: Left ventricular hypertrabeculation/noncompaction with PMP22 duplication-based Charcot–Marie–Tooth disease type 1A. Cardiology 2006; 105: 142–145
28) McMahon CJ, Chang AC, Pignatelli RH, et al: Left ventricular noncompaction cardiomyopathy in association with trisomy 13. Pediatr Cardiol 2005; 26: 477–479
29) Kanemoto N, Horigome H, Nakayama J, et al: Interstitial 1q43-q43 deletion with left ventricular noncompaction myocardium. Eur J Med Genet 2006; 49: 247–253
30) Pauli RM, Scheib-Wixted S, Cripe L, et al: Ventricular noncompaction and distal chromosome 5q deletion. Am J Med Genet 1999; 85: 419–423
31) Luxan G, Casanova JC, Martinez-Poveda B, et al: Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med 2013; 19: 193–201
32) Holmgren D, Wahlander H, Eriksson BO, et al: Cardiomyopathy in children with mitochondrial disease; clinical course and cardiological findings. Eur Heart J 2003; 24: 280–288
33) Scaglia F, Towbin JA, Craigen WJ, et al: Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics 2004; 114: 925–931
34) Yaplito-Lee J, Weintraub R, Jamsen K, et al: Cardiac manifestations in oxidative phosphorylation disorders of childhood. J Pediatr 2007; 150: 407–411
35) Schiff M, Ogier de Baulny H, Lombes A: Neonatal cardiomyopathies and metabolic crises due to oxidative phosphorylation defects. Semin Fetal Neonatal Med 2011; 16: 216–221
36) Papadopoulou LC, Sue CM, Davidson MM, et al: Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat Genet 1999; 23: 333–337
37) Cizkova A, Stranecky V, Mayr JA, et al: TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat Genet 2008; 40: 1288–1290
38) D’Adamo P, Fassone L, Gedeon A, et al: The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am J Hum Genet 1997; 61: 862–867
39) Bates MG, Bourke JP, Giordano C, et al: Cardiac involvement in mitochondrial DNA disease: Clinical spectrum, diagnosis, and management. Eur Heart J 2012; 33: 3023–3033
40) Fayssoil A: Heart diseases in mitochondrial encephalomyopathy, lactic acidosis, and stroke syndrome. Congest Heart Fail 2009; 15: 284–287
41) Anan R, Nakagawa M, Miyata M, et al: Cardiac involvement in mitochondrial diseases. A study on 17 patients with documented mitochondrial DNA defects. Circulation 1995; 91: 955–961
42) Tanaka M, Obayashi T, Yoneda M, et al: Mitochondrial DNA mutations in cardiomyopathy: Combination of replacements yielding cysteine residues and tRNA mutations. Muscle Nerve Suppl 1995; 3 S14: S165–S169
43) Merante F, Tein I, Benson L, et al: Maternally inherited hypertrophic cardiomyopathy due to a novel T-to-C transition at nucleotide 9997 in the mitochondrial tRNA(glycine) gene. Am J Hum Genet 1994; 55: 437–446
44) Schwartz ML, Cox GF, Lin AE, et al: Clinical approach to genetic cardiomyopathy in children. Circulation 1996; 94: 2021–2038
45) Havndrup O, Christiansen M, Stoevring B, et al: Fabry disease mimicking hypertrophic cardiomyopathy: Genetic screening needed for establishing the diagnosis in women. Eur J Heart Fail 2010; 12: 535–540
46) Blair E, Redwood C, Ashrafian H, et al: Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: Evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet 2001; 10: 1215–1220


This page was created on 2018-05-25T14:20:57.243+09:00
This page was last modified on 2018-09-11T17:27:02.59+09:00
このサイトは(株)国際文献社によって運用されています。