主訴
興奮時や運動時の失神
現病歴
発端者は女児.6歳時より興奮時や運動時に意識消失して転倒し,約1分間四肢を強直させるエピソードを3か月間で5回認めたため来院した.発作前に動悸を自覚することがあり,失禁を伴っていることが多かった.
既往歴
特記すべきことなし.来院は学校心臓検診を受ける前であった.
家族歴
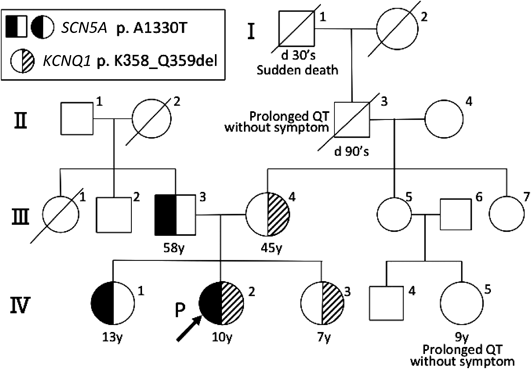
両親および2人の同胞(3歳年上の姉と3歳年下の妹の三姉妹)に失神や痙攣の既往はなく,姉は学校心臓検診でも異常を指摘されていなかった.しかし母方の曽祖父が30歳台で原因不明の突然死をしていた(Fig. 1 I-1).母方の祖父と従兄弟に無症状のQT延長が後に判明した(Fig. 1 II-3, IV-5).聴力障害の家族歴はなかった.
初診時所見および検査
身体所見上は特記すべき異常を認めなかった.まずてんかんを疑い,血液検査(電解質,アンモニア,乳酸ピルビン酸など),脳波,頭部CTを行ったが,異常を認めなかった.
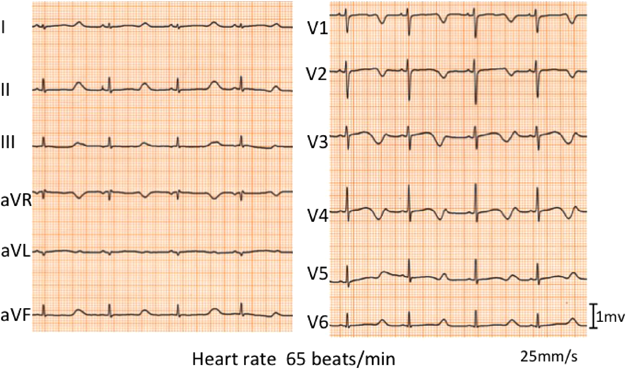
発作前に動悸を伴うことから安静時心電図を確認したところ(Fig. 2),補正QT時間(Bazett)(corrected QT interval: QTcB)=0.67s,補正QT時間(Fridericia)(QTcF)=0.66 sと著明なQT延長を認めた.QT延長の程度が著しいため,診断時の運動負荷心電図は施行しなかった.ホルター心電図では,年齢不相応な徐脈やT wave alternans, TdPは認めず,夜間の徐脈時(QTcB=0.64 s, QTcF=0.65心拍数50/min)にも,活動時(QTcB=0.68 s, QTcF=0.64心拍数88/min)と同様に著明なQT延長を認めた.QT時間の明らかな延長(QTcB>0.48 s)と,ストレスに伴う失神でSchwartz scoreは5点1)であり,以上の検査結果からLQTS確定診断とした.遺伝子検査については両親の同意が得られず施行しなかった.
治療および経過
発端者に対する治療として,atenolol 25 mg/day(1.3 mg/kg/day)内服を開始した.薬剤開始後に運動負荷心電図を施行したが,安静時のQT延長(QTcB=0.65 s, QTcF=0.62)と比較し,運動終了後4分の時点でもQTcB=0.69 s, QTcF=0.66 s(心拍数90/min)と依然著明なQT時間延長を認めていた.運動制限は水泳・マラソン禁止,中等度まで可(管理区分D,運動クラブ活動 禁)とした.
内服開始後2回失神を認めたが,いずれも怠薬によるものと考えられたため服薬指導を行い,以後薬剤内服中の失神は消失した.しかし薬剤開始4年後の10歳時,服薬アドヒアランス良好であったにもかかわらず自転車走行中に失神を認め,治療方針再考を要した.
まずatenololを体重増加とともに増量しておらず,投与量が相対的に減量となっていたため,25 mg/day(0.9 mg/kg/day)から50 mg/day(1.7 mg/kg/day)に増量した.また運動制限を中等度の運動まで(管理区分D)から軽度の運動まで(管理区分C)に強化した.心電図波形ではT波の出現が遅延するLQT3の特徴をもっていたこと(Fig. 2)もあり,Naチャネル遮断薬mexiletine 300 mg/day(10.3 mg/kg/day)を併用した.
さらに著明なQT延長および治療抵抗性からも複合変異症例である可能性も考えられ,今後の治療方針決定に非常に重要であることを家族に再度説明し,同意を得て発端者・両親・姉妹の遺伝子検査を施行した.
遺伝子検査結果とその後の治療
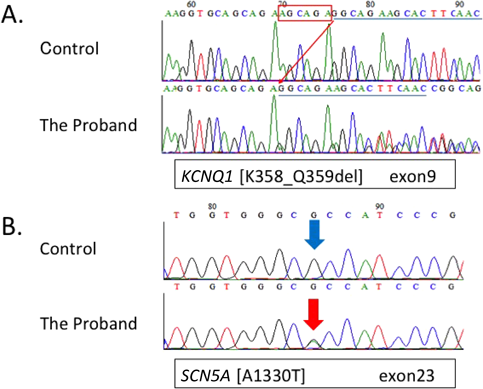
報告されているLQTS原因遺伝子のエクソン領域のスクリーニングを行ったところ,発端者にKCNQ1 c.1073_1078delAGC AGA, p.K358_Q359del(Fig. 3A)と,SCN5A c.3988G>A, p.A1330T(Fig. 3B)の複合変異ヘテロ接合体を同定した.同時に父および13歳の姉にSCN5A変異(Fig. 3B)を,母と7歳妹にはKCNQ1変異(Fig. 3A)を,いずれもヘテロ接合性の変異として認めた.
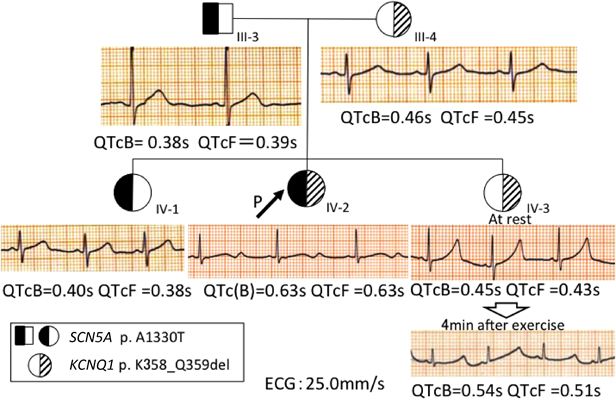
遺伝子検査で判明した変異を踏まえ,症状や臨床検査,治療経過を加味して各人の治療方針を決定した.家人それぞれの心電図をFig. 4に示す.
発端者は複合変異で,QT延長も著明であることより植込み型除細動器(Implantable Cardioverter Defibrillators: ICD)の適応と考えているが,メリットとデメリットを本人および両親に説明して,埋め込み方法や時期を決定していく方針である.現在,β遮断薬とNaチャネル遮断薬の併用および運動制限を継続し,8か月間失神を起こさず経過している.ただしNaチャネル遮断薬開始後もQTcB=0.63 s, QTcF=0.63 sとQTcの短縮は軽度にとどまった(Fig. 4 IV-2).
父と姉に認めたLQT3の遺伝子変異は,データベースにも登録された(reference SNP ID number 199473224)病原性変異である.しかし姉は安静時心電図でQTcB=0.40 s, QTcF=0.38 sとQT延長を認めず(Fig. 4 IV-1),またホルター心電図でも夜間の徐脈時にQTcB=0.45 s, QTcF=0.47 s(心拍数42/min)と軽度のQT時間延長を認めるにとどまった.さらに無症状であり,父の家系に突然死や失神の例がいないことも考慮し,予防的内服はせずに心電図で慎重に経過観察することとした.
一方,母と妹のLQT1の変異には過去に報告はないものの,coding DNAの欠失変異であるため機能異常をきたす可能性が高いと思われた.実際変異をもつ妹はFig. 4 IV-3に示す通り,トレッドミル運動負荷(Bruce法)後の心電図でQTcB=0.54 s, QTcF=0.51 sとQTが延長したため,突然死例を含んでいる母方の家族歴も考慮し一次予防を施行することとした.現在β遮断薬および運動制限(強い運動可,管理区分E,運動クラブ活動 禁,水泳・マラソン・強制運動禁止)で経過観察中である.
両親からそれぞれ別のLQTS責任遺伝子異常を引き継いだ複合変異例である発端者の診断を契機に,両親と同胞の遺伝子異常が明らかとなった一家系を経験した.三姉妹はそれぞれ別の形で変異を引き継いでおり,遺伝子型と表現系を総合的に判断して,個別の治療方針を決定することが重要であった.
まず発端者は父からのLQT3のSCN5A変異と,母からのLQT1のKCNQ1変異を受け継いだ複合変異症例であった.このような複合変異は,遺伝子変異を同定されたLQTS症例の7~8.4%と報告されており,QT延長が著明で,発症年齢が若く,心事故や突然死を起こす確率が高いと言われている2, 3).発端者もQT延長が著明である点,年少時より失神を繰り返した点は複合変異例の特徴を有していたといえる.
失神は運動時にみられ,運動負荷でのQT延長を認めたことや,β遮断薬が治療開始当初は有効と考えられたことでLQT1と診断していたが,心電図波形やβ遮断薬への治療抵抗性から複合変異の可能性が考え,遺伝子検査を積極的に進めた結果,複合変異が明らかとなった.
遺伝子変異が明らかになることで,まず内科治療の妥当性が検討できた.LQT1に対するβ遮断薬の有効性は多数報告されていることに加え,LQT3でも女性においてβ遮断薬の効果が示されており4),増量したβ遮断薬を継続する根拠となった.また日本循環器学会のガイドライン5)では,遺伝子特異的な治療として有症状のLQT3に対するNaチャネル遮断薬の使用はクラスIIaとされており,併用治療を継続する裏づけを見いだせたことは重要であった.
またこのような複合変異症例においてICDは治療の選択肢の一つとなりうる.The Cardiac Society of Australia and New Zealandからのguideline update6)ではQT延長が著明(QTcB>0.55 s)である複合変異例において,一次予防の相対的な適応となっている.また複合変異症例か否かにかかわらず,上記のガイドライン6)とHRS/EHRA/APHRSによるガイドライン7),および本邦のガイドライン5)いずれも,β遮断薬が無効な有症状のLQTSの症例においてICD導入はクラスIIaの推奨となっている.今回の発端者が失神した際にはβ遮断薬の投与量が少なかった可能性があり治療抵抗性と言い切れない部分もあるが,著明なQT延長と複合変異例であることも考慮し,ICD治療は有用であると考えた.
しかし一方でICD治療は合併症が少なくない.不適切作動や,それが原因で生じる不安・鬱などの精神的な有害事象は小児でも報告されている8, 9).また小児ではリード断線含むリード不全の頻度が約10~20%と成人より高い確率で起こると報告されている8, 10).ICDリード留置法の選択は,導入時の体格や両親の身長から予測される留置後の身長変化を考慮して行われるが,経静脈での留置が困難で,より侵襲が大きい心外膜リードを選択せざるを得ないケースも存在する.リード不全は適切なICDの作動を妨げるだけでなく,リード再留置による侵襲もあり,小児のICD導入には大きな問題となる.
上記のように小児期のICD導入には有害事象も少なくないため,ICD導入はメリット・デメリットを本人や保護者が十分に理解してから行う必要がある.本症例ではβ遮断薬とNaチャネル遮断薬で症状が落ち着いていることもあり,現時点では本人および両親はICD治療を希望していない.
父および姉で見つかったLQT3のSCN5A c.3988G>A, p.Ala1330Thrの変異は,NaチャネルのSCN5Aの1330番目のアミノ酸がアラニンからトレオニンに置換されるミスセンス変異である(Reference SNP ID number 199473224).この変異はパッチクランプ法によって,INaチャネルでの第4相の内向き電流が増幅された結果,再分極が遅れるという機能解析がされていることに加え,本変異を有する症例でQT延長に伴う突然死が報告されており11),データベース上も病原性と登録されている.しかしながら,本変異を有する父・姉ともに安静時心電図のQT時間は延長していなかった.HRS/EHRA/APHRSのガイドライン7)やLQT3の多施設研究4)では,心血管イベントのリスクはQT延長の程度と相関するとされており,また父の家系には突然死や失神例がないことや,本人に症状がないことから,現時点では一次予防をせずに経過観察とした.女性の場合は男性と比較し成人期にイベントのリスクが高くなるという報告もあり4)今後も注意深く経過観察していくことが必要である.
母,妹に見つかったLQT1のKCNQ1 c.1073_1078delAGC AGA, p.K358_Q359delの変異は遺伝子の欠失により358番359番のリシン,グルタミンが欠失するインフレームシフト変異である.機能解析の報告はなく,データベース上も病原性についての登録はない(Reference SNP ID number 397508074).よって変異についての考察および臨床症状の両者が治療方針決定に重要となる.
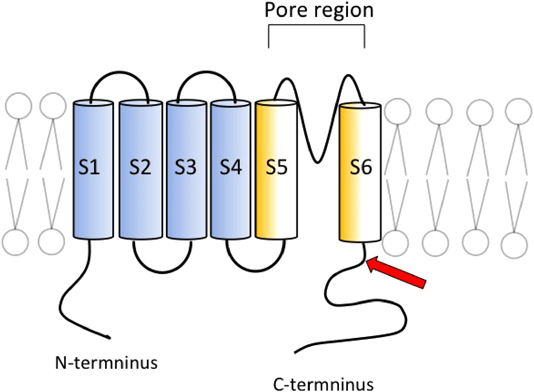
LQT1では遺伝子変異部位や変異タイプが表現型の重症度に及ぼす影響が検討されており12),一般的には膜貫通領域やリンカー領域,およびpore領域の変異で心事故発生が多いと報告されている12, 13).今回の変異の位置は前述の部位ではなく,C末端部位であった(Fig. 5矢印).しかし変異の位置は,立体構造となった場合の詳細な位置関係は不明ではあるが,アミノ酸配列ではpore部位(285–355番)に近接した2アミノ酸の脱落であり,この変異の病的意義は否定できない.また変異タイプも重要であり,このような非リピート領域のインフレームシフトはAmerican College of Medical Genetics and Genomicsのガイドライン14)によると,病原性のエビデンスは中等度とされている点からも,この変異の病原性が裏づけられる.
さらにKCNQ1のヘテロ変異接合体では,蛋白構造変化が軽微であったとしてもチャネル機能が健常者の1/2以下となる,ドミナントネガティブ型抑制を取ることが多い12, 13).KCNQ1(Fig. 5)は四量体蛋白であり,変異型サブユニットが一つ以上含まれる機能異常を有するチャネルが,全体の15/16形成されることとなるため,一般的に表現型が極端になる.
以上変異の部位がpore領域に近いこと,非リピート領域のインフレームシフトであること,KCNQ1の変異がドミナントネガティブ型抑制をとることから本変異は病原性が高いと判断した.実際妹は運動負荷後にQT延長を認め,母方の家族歴に突然死があること(Fig. 1 I-1)も含め,遺伝型・表現型双方より考慮し病原性変異と考え,βブロッカーおよび運動制限による一次予防を開始した.今後機能解析が進めば本遺伝子異常の病的意義がより明らかになると思われる.
今回の遺伝子検査により姉妹それぞれに別の変異が見つかり,臨床経過も様々であった.発端者では複合変異が見つかったことが内服の治療強化を進める根拠となり,ICDも検討することとなった.姉ではLQT3の病原性変異と報告されている異常を認めた.ただしQT延長は軽度で表現型では軽症と考えられたため経過観察のみとした.妹のもつLQT1の変異は病的意義が不明ではあったが,遺伝子変異部位や変異タイプの検討と遺伝子変異がドミナントネガティブ型抑制となりやすいこと,および運動負荷心電図でのQT延長や家族歴の臨床的な観点も含め考察し,一次予防を施行した.
上記のように三姉妹において遺伝子異常や表現型は三者三様であり,遺伝子異常もしくは表現型どちらかのみでの治療方針決定は困難であった.遺伝子検査は有効な治療法や生活指導などを知ることができる非常に有用な検査であるが,遺伝子異常の病原性に対する考察を行い,臨床検査や家族歴を含む表現型も十分に考慮したうえでの治療方針決定が重要と考えられた.