ミトコンドリア心筋症Mitochondrial Cardiomyopathy
北海道大学病院小児科Department of Pediatrics, Hokkaido University Graduate School of Medicine ◇ Hokkaido, Japan
発行日:2017年7月1日Published: July 1, 2017
ミトコンドリア心筋症は,ミトコンドリアの構造,機能に関わる遺伝子の異常によって生じる酸化的リン酸化障害を特徴とする心筋症である.通常は全身のミトコンドリア病の心合併症として認識されることが多いが,心筋症が唯一の症状である場合には見逃されることが多い.原因遺伝子はミトコンドリアDNAと核遺伝子に存在し,症状は無症状のものから重症心不全,突然死まで様々である.近年,網羅的遺伝子検査の発達によりミトコンドリア病の原因遺伝子が次々と同定され始めてきているが,ミトコンドリア病は本来罹患組織の酸化的リン酸化障害を証明することで確定診断されてきた経緯があり,組織の生化学的検査は今なお避けては通れない検査として位置づけられている.
Mitochondrial cardiomyopathy is characterized by an oxidative phosphorylation (OXPHOS) disorder due to genetic mutations in genes encoding the structure and function of myocardial mitochondria. Mutations in both mitochondrial and nuclear DNA can cause mitochondrial cardiomyopathy, and it is typically recognized as one of the generalized manifestations of neurological and metabolic disorders. Cardiomyopathy, however, can be the only phenotype of mitochondrial disease and is often misdiagnosed. Cardiac manifestations vary from asymptomatic to catastrophic heart failure or sudden death. Although recent evolution in genetic tests has allowed the identification of the causative gene, tissue sampling to identify an OXPHOS disorder is still regarded as the gold standard for diagnosis.
Key words: mitochondrial cardiomyopathy; oxidative phosphorylation (OXPHOS); mitochondrial DNA; respiratory chain complex
© 2017 特定非営利活動法人日本小児循環器学会© 2017 Japanese Society of Pediatric Cardiology and Cardiac Surgery
ミトコンドリア心筋症は,ミトコンドリアの構造,機能に関わる遺伝子の異常によって生じる酸化的リン酸化障害を特徴とする心筋症である1).
ミトコンドリア心筋症はミトコンドリア病の一症状として認識されていることが多く,乳児期の筋力低下や代謝性アシドーシスに伴う重症心不全,あるいは,MELASやLeigh脳症などのミトコンドリア病に合併する心筋症というイメージが強いかと思われる.孤発性の心筋症の中では希少であり,ミトコンドリア病の随伴症状がなければ見逃されやすい.近年,次世代シーケンサーを用いた網羅的遺伝子検査による新規原因遺伝子の同定や酸素消費速度測定による細胞単位でのミトコンドリア機能評価法が診断に応用されるようになり,ミトコンドリア心筋症の診断がより精密かつ簡便になりつつあることも事実である.また,最近の研究では不全心筋においてミトコンドリアが大きな役割を果たしていることも明らかにされてきており,ミトコンドリア心筋症を学ぶことは心筋エネルギー代謝に関わる遺伝子や酵素,それらの評価系を学ぶことに繋がる.今回はミトコンドリアの正常な構造と機能,ミトコンドリア心筋症の分子遺伝学,病態と臨床像,および確定診断について解説する.
ミトコンドリアはほとんど全ての真核生物に存在する細胞小器官(オルガネラ)で,好気呼吸によって組織に必要なATPを産生する.心筋は成人で1日に30 kgのATPを消費する臓器であり,1つの心筋細胞にはミトコンドリアが数百個と豊富に存在する.ミトコンドリアは外膜と内膜より作られ,内膜はクリステと呼ばれるひだを形成して4つの呼吸鎖酵素複合体(respiratory chain complex; RC, Complex I~IV)で構成される電子伝達系とATP合成酵素(Complex V)が存在している.内膜にはカルジオリピンという複合脂質が豊富に含まれ,膜不透過性と呼吸鎖酵素複合体の安定化に大きな役割を果たしている.内膜の内部であるマトリックスに運ばれた脂肪酸などの基質からβ酸化およびTCA回路によりH+の供給源であるNADH, FADH2が生成され,RCによる酸化でH+が膜間腔に汲み出され,内膜を隔てたプロトン勾配によってATP合成酵素の存在下にATPを合成する.同時に発生した電子は電子伝達系(コエンザイムQ-Complex III-チトクロームc)に渡り最終的に酸素分子を水に還元する(Fig. 1).
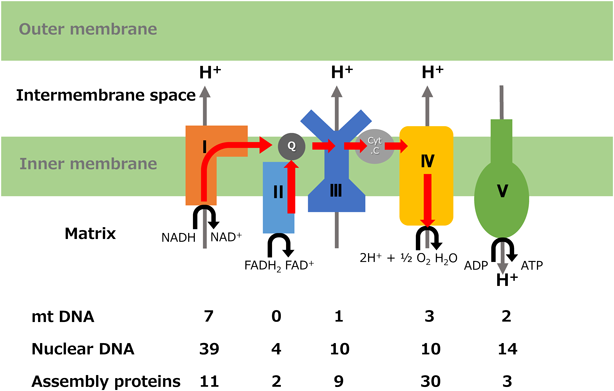
ミトコンドリアの構造と機能の維持には約1,500個の蛋白が関与しているが,ミトコンドリアDNA(mtDNA)でコードされているものはそのうちわずか1%未満で残りの99%は核遺伝子でコードされている.mtDNAはミトコンドリア内に独自に存在する16569塩基の環状DNAで1つの心筋細胞に数千コピー存在し,RC(Complex IIを除く)の13個のサブユニット(複合体を構成している蛋白群)をコードしているほか,翻訳や合成に関わる22個のtRNAと2個のrRNAをコードしている(Fig. 2)2).核遺伝子は残りのRCサブユニットやそれらを組み立てるためのアセンブリ因子,mtDNAのメンテナンス,ミトコンドリアへの輸送に関わる蛋白などをコードし,細胞質内で蛋白を合成してミトコンドリア内に輸送している.mtDNAの複製にはDNAポリメラーゼγ(POLG)やmtDNAヘリカーゼとして機能するTwinkle蛋白が関与し,mtDNAの転写,翻訳にはミトコンドリアRNAポリメラーゼ(mtRNAP),ミトコンドリア転写因子(TFAM)および翻訳伸長因子(TSFM)が関与するが,これらは全て核遺伝子にコードされている.
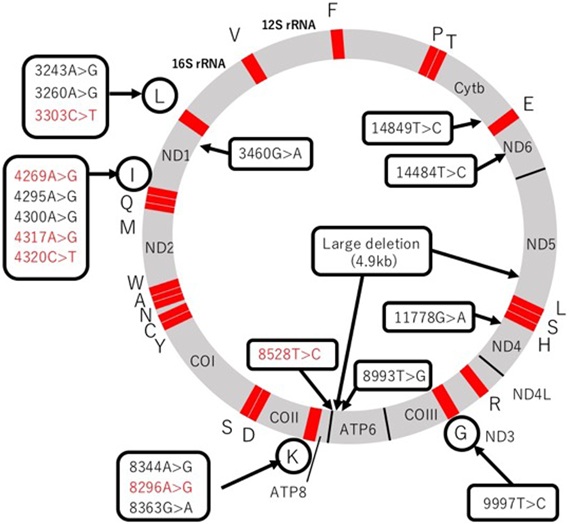
Individual capital letters represent each tRNA. Mutations in red indicate early onset cardiomyopathy during infancy. ND1, ND2, ND3, ND4L, ND4, ND5, ND6: Complex I subunit genes. Cytb: Complex III subunit gene. COI, COII, COIII: Complex IV subunit genes. ATP6, ATP8: Complex V subunit genes.
ミトコンドリア心筋症の原因遺伝子にはmtDNA変異と核遺伝子変異が存在し,これらの遺伝学的背景は大きく異なるが,心筋症の表現型は類似しており,臨床経過や症状のみからではこれらの鑑別は難しい.
ミトコンドリアの構造,機能に関わるタンパクをコードしている遺伝子にはミトコンドリアDNA(mtDNA)と核遺伝子があるが,mtDNAは酸化的リン酸化(oxidative phosphorylation; OXPHOS)に関わる遺伝子のわずかしか関与しておらず,ミトコンドリア病のほとんどが核遺伝子変異によることが判明している.mtDNAの異常には点変異と欠失が存在するが,いずれの場合でも正常型と変異型のmtDNAが混在する状態(ヘテロプラスミー)が存在し,ある一定の変異率を超えることで症状が発現する(閾値効果).また,この閾値効果のため年齢を経ないと発症しないことがあり,mtDNA変異によるミトコンドリア病が成人に多く,小児期発症は核遺伝子変異に多いことが知られている.
また,臓器・組織によっても変異率や閾値が異なる(組織特異性)ため,mtDNA変異を確定診断するには罹患臓器のmtDNA変異と変異率を考慮することが必要である3).たとえば,血液からのmtDNA抽出においてmtDNA変異率が極めて低い場合でも心筋では変異率の高いmtDNA変異が存在している可能性がある.
受精卵においてmtDNAは卵母細胞にのみ存在し,母性遺伝という独特の遺伝形式を有する.変異率は同胞間においても個人差が大きく発症年齢や重症度は家系間においても様々であり,遺伝カウンセリングは慎重に進めなければならない4).mtDNAの欠失はKearns–Sayre症候群に特徴的で心合併症としては心伝導障害が起こることが知られている.核遺伝子変異ではこれまでに250種以上のミトコンドリア病原因遺伝子が報告されており,遺伝形式は常染色体優性遺伝,常染色体劣性遺伝,X連鎖性劣性遺伝など全てのパターンがある.出生前診断においてmtDNA変異のミトコンドリア異常の解釈はヘテロプラスミーのため困難である一方,核遺伝子変異で致死的な疾患では遺伝カウンセリングが重要である.
ミトコンドリア心筋症関連遺伝子と表現型をTable 1に示す5).
| I.ミトコンドリアDNA | 遺伝子 | 変異 | 心筋症 | 表現型(OMIM) |
|---|---|---|---|---|
| 呼吸鎖複合体サブユニット | ||||
| MT-ND4 | m.11778G>A | HCM | LHON/Progressive Dystonia | |
| MT-ATP6/8 | m.8528T>C | HCM | Infantile cardiomyopathy | |
| MT-ATP6 | m.8993T>G | HCM | NARP/Leigh Disease | |
| MT-ND6 | m.14484T>C | DCM | LHON | |
| MT-CYB | m.14849T>C | HCM | Septo-Optic Dysplasia | |
| ミトコンドリア蛋白合成 | ||||
| MT-TL1 | m.3243A>G | HCM, DCM, RCM, LVNC | MELAS/Leigh Syndrome/CPEO/Mitochondrial Mitopathy | |
| m.3260A>G | HCM, DCM | MELAS/Maternal Myopathy and Cardiomyopathy | ||
| m.3303T>C | HCM, DCM | Maternal Myopathy and Cardiomyopathy | ||
| MT-TI | m.4300A>G | HCM, DCM | Maternally Inherited Cardiomyopathy | |
| MT-TK | m.8344A>G | HCM, DCM | MERRF | |
| m.8363G>A | HCM, DCM | MERRF/Leigh Syndrome | ||
| MT-RNR1 | m.1555A>G | RCM | Maternally inherited DEAFness | |
| II.核遺伝子 | 遺伝子 | OMIM ID | 心筋症 | 表現型(OMIM) |
| 呼吸鎖複合体サブユニット | ||||
| 複合体I | NDUFS2 | 252010 | HCM | Mitochondrial complex I deficiency |
| 複合体I | NDUFV2 | 252010 | HCM | Mitochondrial complex I deficiency |
| 複合体I | NDUFA11 | 252010 | HCM | Mitochondrial complex I deficiency |
| 複合体II | SDHA | 252011 | DCM, LVNC | Mitochondrial complex II deficiency |
| 複合体アセンブリ | ||||
| 複合体I | NDUFAF1 | 252010 | HCM | Mitochondrial complex I deficiency |
| 複合体I | ACAD9 | 611126 | HCM | Mitochondrial complex I deficiency due to ACAD9 deficiency |
| 複合体IV | SCO2 | 604377 | HCM | CEMCOX1 (fatal infantile cardioencephalomyopathy due to cytochrome c oxidase (COX) deficiency 1) |
| 複合体IV | COX10 | 220110 | HCM | Mitochondrial complex IV deficiency |
| 複合体IV | COX15 | 615119 | HCM | CEMCOX2 (fatal infantile cardioencephalomyopathy due to cytochrome c oxidase (COX) deficiency 2) |
| 複合体V | TMEM70 | 614052 | HCM | MC5DN2 (mitochondrial complex V (ATP synthase) deficiency nuclear type 2) |
| ミトコンドリア蛋白合成 | ||||
| AARS2 | 614096 | HCM | COXPD8 (combined oxidative phosphorylation deficiency-8) | |
| MRPS22 | 611719 | HCM | COXPD5 (combined oxidative phosphorylation deficiency-5) | |
| TSFM | 610505 | HCM | COXPD3 (combined oxidative phosphorylation deficiency-3) | |
| GTPBP3 | 616198 | HCM, DCM | COXPD23 (combined oxidative phosphorylation deficiency-23) | |
| MTO1 | 614702 | HCM | COXPD10 (combined oxidative phosphorylation deficiency-10) | |
| ELAC2 | 615440 | HCM | COXPD17 (combined oxidative phosphorylation deficiency-17) | |
| ミトコンドリア整合性維持 | ||||
| TAZ | 302060 | DCM, LVNC | BTHS (Barth Syndrome) | |
| AGK | 212350 | HCM | Sengers Syndrome | |
| SLC22A5 | 212140 | HCM, DCM | CDSP (Systemic primary carnitine deficiency) | |
| ACADVL | 201475 | HCM, DCM | VLCAD deficiency | |
| HADHA | 609015 | DCM | HADHA (Trifunctional protein deficiency alpha subunit) | |
| ミトコンドリアDNA安定性 | ||||
| SLC25A4 | 615418 | HCM | MTDPS12 (mitochondrial DNA depletion syndrome-12) | |
| 鉄恒常性 | ||||
| FXN | 229300 | HCM | FRDA1 (Friedreich ataxia) | |
| BOLA3 | 614299 | HCM | MMDS2 (multiple mitochondrial dysfunctions syndrome-2)with hypoglycinemia | |
| コエンザイムQ10生合成 | ||||
| COQ9 | 614654 | HCM | COQ10D5 (coenzyme Q10 deficiency-5) | |
| COQ4 | 616276 | HCM | COQ10D7 (coenzyme Q10 deficiency-7) | |
| ミトコンドリア蛋白輸送 | ||||
| DNAJC19 | 610198 | DCM, LVNC | MGCA5 (3-methylglutaconic aciduria type V) | |
| Modified from Ref. 5). DCM; dilated cardiomyopathy, HCM; hypertrophc cardiomyopathy, RCM; restrictive cardiomyopathy, LVNC; left ventricular noncompaction. | ||||
ミトコンドリア心筋症を来す遺伝子を機能的に分類すると,1)呼吸鎖複合体サブユニット,2)複合体アセンブリ,3)ミトコンドリア蛋白合成,4)ミトコンドリア整合性維持,5)ミトコンドリアDNA安定性などに分類される.
呼吸鎖複合体サブユニットの異常によるものはmtDNA変異ではComplex I, Complex IV,およびComplex VのサブユニットをコードするmtDNA変異6)が報告されている.核遺伝子変異ではComplex Iサブユニットをコードする遺伝子変異で肥大型心筋症(HCM)を来すものにNDUFS2, NDUFV2, NDUFA11が報告されており,いずれもLeigh脳症の原因遺伝子として知られている7–9).Complex IIサブユニットをコードする遺伝子変異ではSDHAが報告されており,拡張型心筋症(DCM),左室心筋緻密化障害(LVNC)を来し,乳児発症例では多くが致死性である10).Complex Iの複合体の集合に関わるアセンブリファクターの異常にはLeigh脳症が多く,その中でNDUFAF1, ACAD9は心筋症の合併が認められている11, 12).Complex IVのアセンブリファクターではSURF1が有名であるが,心筋症ではSCO2, COX10, COX15が重要である13–15).Complex VではTMEM70が早期のComplex Vアセンブリに必要な因子で,この部位の変異は乳児発症の乳酸アシドーシス,3-メチルグルタコン尿症,および心筋症を来すことが知られている16).ミトコンドリア蛋白合成に関わる遺伝子はmtDNAではtRNAをコードする遺伝子異常があり,m.3243A>G変異はMELASの原因遺伝子として有名である.核遺伝子変異ではtRNAの翻訳後修飾やアミノアシルtRNA合成酵素(aaRS)の異常が知られ,心筋症ではAARS2変異が乳児発症HCMの原因遺伝子として報告されている17, 18).ミトコンドリア整合維持に関わる遺伝子はカルジオリピンの成熟に関わる酵素tafazzinをコードする遺伝子TAZが知られ,Barth症候群の責任遺伝子として知られている.心合併症にはLVNCが多く,男児の乳児重症心不全の鑑別診断に極めて重要な遺伝子である19).その他,ミトコンドリアにおけるリン脂質代謝異常ではAcylglycerol Kinaseの欠損(AGK変異)による心筋症や白内障を特徴としたミトコンドリア病(Sengers症候群)が知られている20).ミトコンドリアDNAの安定に関わる遺伝子ではSLC25A4がmtDNA枯渇症候群の原因遺伝子として知られ,これらの変異により心筋症を来す21).
ミトコンドリア心筋症に共通の病態は単位心筋ミトコンドリアにおけるATP産生能の低下である.心筋は持続的に好気的代謝によってエネルギーを消費し続ける臓器であり,ATPの欠乏は心筋の収縮性低下に直結する22).胎児心筋は低酸素環境下でミトコンドリアによる好気性代謝は抑えられており,ミトコンドリアに機能障害があっても顕性化していない場合がある.生後はエネルギーを好気性代謝に依存するため,時に新生児・乳児期に重篤な心筋症として発症する場合は心筋ミトコンドリアの適応不全が存在している可能性がある23).乳児以降に発症するミトコンドリア心筋症ではATP産生能の低下を代償してミトコンドリアが心筋細胞内に充満しており,筋原線維の偏位や活性酸素種の増加が認められている.これらは将来的にミトコンドリアのさらなる質的低下を招いて最終的に心筋不全に至るものと考えられている.臨床では全周性のHCMから拡張相HCMへの移行として認められることが多い.その他,LVNCや,心伝導障害,肺高血圧などの表現型が報告されているが,ミトコンドリア機能とこれらの疾患における詳細なメカニズムは明らかにされていない.
ミトコンドリア心筋症は無症状で経過するものから重篤な心不全に至るまで重症度は様々であり,時に致死性不整脈や突然死を来すこともある.感染などの生体へのストレスを契機に悪化することがあり,診断のきっかけになる場合もある.多くは神経筋疾患や代謝性疾患などのミトコンドリア病の随伴症状を伴うが,心筋症孤発例や心筋症がミトコンドリア病診断のきっかけになる場合もある.代表的な表現型はHCM, DCM, RCM, LVNCである.HCMはミトコンドリア心筋症の約半数を占め,ほとんどが全周性の心肥大であるが,閉塞性HCMの報告もみられる.HCMの多くは収縮機能不全から代償不全となり拡張相HCMへ移行することが多い.DCMはHCMについで多く,KSS, MELAS, MERRF, Leigh脳症においても合併することがある.孤発例ではComplex II欠損に認められる.16歳までの生存率は18%との報告があり24),きわめて予後不良である.RCMは稀であるが予後不良であり,心臓移植を施行したミトコンドリア心筋症24例のうち12%がRCMであったとの報告がある25).LVNCの頻度は稀とされているが,FinstererらはLeft ventricular hypertrabeculation(LVHT)/Noncompactionとして報告した187例のうちミトコンドリア心筋症が40例と最も多く,ついでBarth症候群が30例と報告しており,実際には多く存在している可能性がある26).Histiocytoid cardiomyopathy(Purkinje fiber dysplasia)として知られている心筋症は乳児期に心室性不整脈で発症することがあるが,ミトコンドリア病の心病変の一つであることが報告されている27).
不整脈は単独,あるいは上記心筋症に合併して発症する.洞機能不全症候群はKSSやミトコンドリアDNA枯渇症候群に認める.完全房室ブロックはKSSに高率に合併し,ペースメーカ導入のタイミングに留意する.WPW症候群はMELAS, MERRFに多く合併し,上室性頻拍症例ではアブレーションの適応となる場合がある28).ミトコンドリア心筋症は新生児,乳児の肥大型心筋症の鑑別疾患として重要であり,筋疾患や代謝性疾患,難聴などの随伴疾患の可能性を考慮して後述する方法で確定診断を進めていく.ときに経過中に完全房室ブロックや心室頻拍を契機に死亡するケースがあることに留意する.
代表的なミトコンドリア病(MELAS, MERRF, KSS, LHON, Leigh脳症)において心筋症,不整脈を発症することがある.MELASでは40%程度にHCMの合併を認め29),mt.3243 A>G変異率と心肥大の程度に正の相関があることが知られている30).進行例では拡張相HCMへ移行することが多い.MERRFでは半数程度に心筋症の合併を認め,HCM, DCMを来すことが知られている31).KSSでは高度房室ブロックによる失神が45%,突然死が23%に認めており,二枝ブロックの出現で予防的ペースメーカを導入することが勧められている32).LHON, Leigh脳症ではHCMの合併が報告されているが頻度は明らかではない.
ミトコンドリア心筋症確定診断のためのフローチャートをFig. 3に示す5).ミトコンドリア心筋症の確定診断は冒頭の定義にあるように,酸化的リン酸化障害の存在を心筋で証明し,原因遺伝子を証明することである.酸化的リン酸化障害の診断はBernierらによるミトコンドリア呼吸鎖複合体異常症の診断基準(Table 2)が広く用いられており,神経筋疾患領域で広く臨床応用されている33).本診断基準をみるとわかるように,心筋症単独の場合には心筋組織を使用した酵素活性診断を行う必要がある.また,診断基準にも記されているように心筋の超微形態像でミトコンドリア異常を認めた場合も本症を疑うきっかけとなる.しかし,電子顕微鏡像におけるミトコンドリア心筋症の診断基準は特に定められておらず,ミトコンドリア異常増殖,クリステの変性(同心円状,束状),グリコーゲン顆粒の集積といった特徴から疑われることが多い.光顕では空胞変性を認めることが多い.
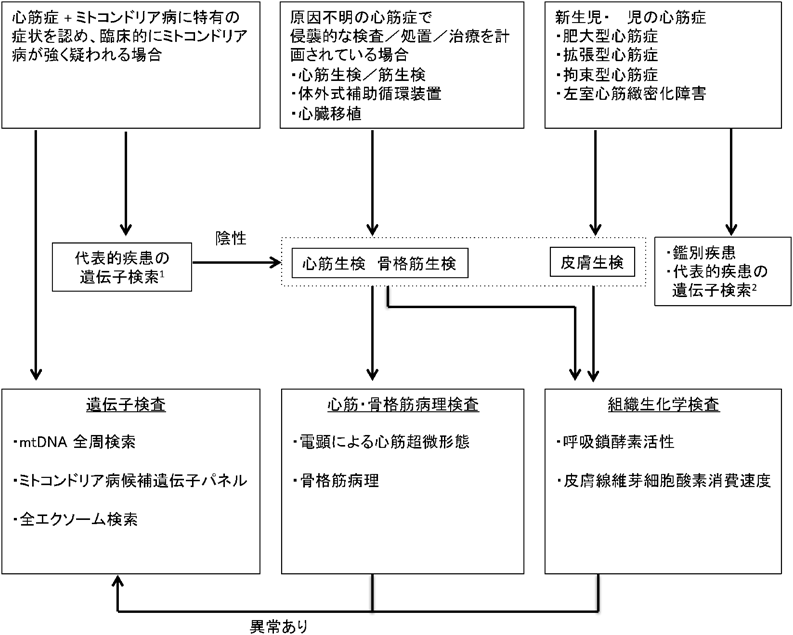
Genetic tests for common mitochondrial diseases: MELAS (mt3243A>G, mt3271T>C), MERRF (mt8344A>G), Leigh encephalopathy (mt8993T>G), KSS/CPEO (mtDNA deletion). Differential diagnosis: Pompe disease, Noonan syndrome, organic acidemia and fatty acid oxidation disorders, sarcomeric gene mutations, and Barth syndrome (TAZ). Modified from Ref. 5).
| ■大基準 |
| I. 臨床症状 |
| 以下の3項目(①~③)をすべて満たす臨床的に確定診断されたミトコンドリア脳筋症またはミトコンドリアサイトパチー |
| ①他の病因では説明できない多臓器にまたがる症状が存在する,少なくとも以下の3系統以上の臓器にまたがること. |
| 1)神経系,2)筋肉,3)心臓,4)腎臓,5)消化器,6)肝臓,7)内分泌,8)造血器,9)耳科,10)眼科,11)皮膚科,12)奇形症候群 |
| ②発作性進行性経過:しばしば感染を機に増悪する,または母系遺伝を思わせる家族歴:母方の親戚の1人以上にミトコンドリア呼吸鎖異常症の疑い例または可能性が存在する |
| ③代謝性あるいは非代謝性の除外診断 |
| II. 病理組織像 |
| 骨格筋2%以上のragged-red fiber |
| III. 酵素活性 |
| ①抗体染色:COX(−)fiber |
| 50歳以下の場合2%以上 |
| 50歳以上の場合5%以上 |
| ②In vitro呼吸鎖酵素活性 |
| 1つの臓器で20%以下,または2つ以上の臓器にまたがって30%以下 |
| 1つの培養細胞で30%以下 |
| IV. 機能解析 |
| 線維芽細胞のATP合成能:平均マイナス3SD以下 |
| V. 分子生物学 |
| 核またはミトコンドリアの明らかな病原遺伝子異常が見つかること |
| ■小基準 |
| I. 臨床症状 |
| 1つでもミトコンドリア呼吸鎖異常症に合致した症状があること |
| II. 病理組織像 |
| ・骨格筋:ragged-red fiber |
| 30~50歳:1~2% |
| 30歳未満:少しでもあればよい |
| ・16歳未満で2%以上の筋線維膜下のミトコンドリア集積 |
| ・臓器は問わずミトコンドリア電顕的異常 |
| III. 酵素活性 |
| ①抗体染色による呼吸鎖酵素欠損の証明 |
| ②In vitro呼吸鎖酵素活性 |
| 1つの臓器で20~30%,または2つ以上の臓器にまたがって30~40% |
| 1つの培養細胞で30~40% |
| IV. 機能解析 |
| 線維芽細胞のATP合成能:平均マイナス2–3SD |
| ガラクトース培地中で成育できない線維芽細胞 |
| V. 分子生物学 |
| 核またはミトコンドリアの病原遺伝子異常の可能性があること |
| VI. 呼吸鎖異常を示唆する1つ以上の検査所見 |
| ①血中,髄液中乳酸・ピルビン酸・アラニン高値 |
| ②髄液中タンパクの増加(KSS疑いの時) |
| ③31P-MRSまたはPETの異常所見(筋肉or脳) |
| ④エルゴメーター異常所見(VO2max, AVO2D,乳酸閾値の低下) |
| Definite:大基準2つ,もしくは大基準1つ+小基準2つ |
| Probable:大基準1つ+小基準1つ,もしくは小基準3つ |
| Possible:大基準1つ,もしくは小基準2つ(うち1つは臨床症状) |
| Modified from Ref. 33). |
原因不明の突然死など剖検例においても何らかの心筋症が疑われた場合に心筋組織の呼吸鎖酵素活性を測定することが有用である.生検,剖検いずれの場合においても採取した組織の一部は必ず未処理のまま−80度で凍結保存して酵素活性を温存しておくことに留意する.ホルマリン(ホルムアルデヒド)水溶液は光顕用のみに使用される固定液であるが「ミトコンドリア心筋症の光顕像は特異的所見に乏しく,摘出心や組織を全てホルマリンに浸してしまうと確定診断が不可能になる」恐れがあるので留意すべきである.
電顕用は最初からグルタールアルデヒド水溶液に浸すことが重要である.仮に電顕用に組織が固定されていなかった場合,ホルマリン固定の段階であれば電顕試料作製に進めていくことは可能であるが,パラフィン包埋ブロックからの電顕試料作成は劣化が著しく診断困難となる.
新生児,乳児の原因不明な心筋症に確定診断の第一段階から心筋生検を行うことはなく,ミトコンドリア病の随伴症状があれば遺伝子検査から進めていくことになる.代表的なミトコンドリア病の遺伝子検査で異常を認めない場合には次世代シーケンサーを用いた全エクソーム検索を行うが,最近は検査の迅速化,効率化を目的として核遺伝子とミトコンドリアDNAを搭載したミトコンドリア病候補遺伝子パネルを用いた遺伝子診断法が始まっている(Fig. 4).

筋力低下などの骨格筋症状があれば骨格筋生検からのアプローチも可能である.Fig. 5にHCMの鑑別診断において軽度の筋力低下を認め筋生検と心筋生検によってミトコンドリア心筋症と診断しえた自験例を示す.本症例では,上記診断基準の大基準2つを満たし(骨格筋Ragged-red fiberの存在,心筋での呼吸鎖酵素活性低下),確定診断となる.
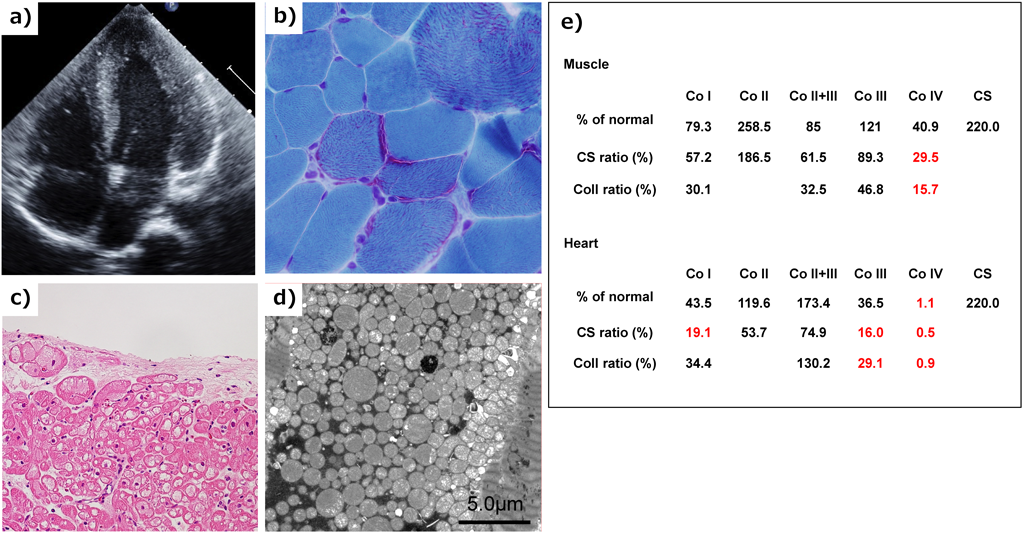
a) Echocardiography showed mild hypertrophic cardiomyopathy. b) Skeletal muscle biopsy with modified Gomori trichrome staining showed ragged-red fibers. c) Light microscopy of a biopsied right ventricle showed cytoplasmic vacuolization in the myocardium. d) Electron microscopy of a biopsied right ventricular endomyocardium showed marked proliferation of mitochondria within myofibrils. e) Respiratory chain complex activity of biopsied skeletal muscle and myocardium showed defects in complex IV and combined defects in Complex I, III, and IV. Co I, Co II, Co II+III, Co III, Co IV: Enzymatic activity of respiratory chain complex in complex I, II, II+III, III, and IV. CS: citrate synthase activity.
最近では皮膚生検から線維芽細胞を作成し,酸素消費速度を測定することで酸化的リン酸化障害を証明する方法が進んでいる34).
一方で前述のように関連遺伝子の種類が非常に多く,診断のハードルが高いのが現状である.今後は遺伝子検査を軸として侵襲度の低いモダリティによって診断精度を高めていくことが要求されるが,希少疾患であるため症例の蓄積が極めて重要であり,診療ネットワークとレジストリシステムの構築が急務である.
ミトコンドリア心筋症に対する根本治療は現在確立したものはなく,一般的には慢性心不全に準じた内科管理を行う.レニン・アンギオテンシン系阻害薬やβ遮断薬における本症の予後改善効果は知られていないが,拡張型心筋症,拡張相肥大型心筋症などの心機能低下例には導入を考慮すべきである.ただし,レニン・アンギオテンシン系阻害薬は腎機能障害がある場合に留意が必要である.ミトコンドリアサイトパチーの際に生じる急性心不全増悪に対してはビタミンカクテル療法に加え,うっ血の改善を目的に利尿薬,PDEIII阻害薬の静脈内投与を行う.心室頻拍,心室細動などの致死性不整脈で発症することもあり,電気的除細動やアミオダロン,Ia群薬を考慮する.ミトコンドリア心筋症の致死性不整脈の発症前予測は困難であり,突然死の予防を目的とした一次予防としてのICD植込術は行われていないが,発症後の二次予防としてのICD植込術は適応となる.心室補助人工心臓(ventricular assist device; VAD)治療については心臓移植が前提となるが,ミトコンドリア病は随伴症状を伴うことが多いため適応になりにくく,他臓器の重症度も鑑みて慎重に検討すべきである.
平成27年よりミトコンドリア病診療の質を高める,レジストリシステムの構築,診断基準・診療ガイドラインの策定および診断システムの整備を行う臨床研究(研究代表 村山 圭)がスタートし,診療マニュアル策定およびレジストリシステムの整備を進めており,診療マニュアルは「ミトコンドリア病診療マニュアル2017(診断と治療社)」として刊行されたのでご参照いただければ幸いである.レジストリシステム(責任者 大竹 明)はMO-Bank(Mitochondrial disease research Organization data Bank)(HP: http://mo-bank.com/index.html)を軸に,ミトコンドリア病の国際的なレジストリシステムであるGlobal Mito Registryへの協力体制も視野に整備されてきている.
最後に,現在ミトコンドリア心筋症に特異的な治療法はなく,心保護療法や対症療法を行っているのが現状である.原因遺伝子によっても治療が異なってくるのは必然であるし,薬剤がミトコンドリア内膜を通過しにくいことも最大の難点とされている.しかしながら病気の進行過程には活性酸素種の増加に伴うミトコンドリア障害など共通のpathwayも存在しており今後治療のターゲットになりうるものと思われる.また,ミトコンドリアマトリックスへの薬物送達研究が最近注目されてきており,今後大きなパラダイムシフトが起こることを期待したい.
稿を終えるにあたり,呼吸鎖酵素活性の資料をご提供頂いた千葉こども病院代謝科 村山圭先生,ミトコンドリア病遺伝子パネルをご提供頂いた,埼玉医科大学ゲノム医学研究センタ所長 岡崎康司先生,同 小児科 大竹明先生,筋病理をご提供頂いた楡の会こどもクリニック 須藤章先生に深謝致します.
本論文について,開示すべき利益相反(COI)はない.
1) Meyers DE, Basha HI, Koenig MK: Mitochondrial cardiomyopathy: Pathophysiology, diagnosis, and management. Texas Heart Institute journal/from the Texas Heart Institute of St Luke’s Episcopal Hospital. Texas Children’s Hospital 2013; 40: 385–394
2) Schapira AH: Mitochondrial disease. Lancet 2006; 368: 70–82
3) Rossignol R, Faustin B, Rocher C, et al: Mitochondrial threshold effects. Biochem J 2003; 370: 751–762
4) Vento JM, Pappa B: Genetic counseling in mitochondrial disease. Neurotherapeutics 2013; 10: 243–250
5) 日本ミトコンドリア学会(編),村山 圭,小坂 仁,米田 誠:ミトコンドリア病診療マニュアル2017.東京,診断と治療社 2016, pp 69–76
6) Imai A, Fujita S, Kishita Y, et al: Rapidly progressive infantile cardiomyopathy with mitochondrial respiratory chain complex V deficiency due to loss of ATPase 6 and 8 protein. Int J Cardiol 2016; 207: 203–205
7) Loeffen J, Elpeleg O, Smeitink J, et al: Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann Neurol 2001; 49: 195–201
8) Benit P, Beugnot R, Chretien D, et al: Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum Mutat 2003; 21: 582–586
9) Berger I, Hershkovitz E, Shaag A, et al: Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann Neurol 2008; 63: 405–408
10) Levitas A, Muhammad E, Harel G, et al: Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur J Hum Genet 2010; 18: 1160–1165
11) Fassone E, Taanman JW, Hargreaves IP, et al: Mutations in the mitochondrial complex I assembly factor NDUFAF1 cause fatal infantile hypertrophic cardiomyopathy. J Med Genet 2011; 48: 691–697
12) Leslie N, Wang X, Peng Y, et al: Neonatal multiorgan failure due to ACAD9 mutation and complex I deficiency with mitochondrial hyperplasia in liver, cardiac myocytes, skeletal muscle, and renal tubules. Hum Pathol 2016; 49: 27–32
13) Leary SC, Mattman A, Wai T, et al: A hemizygous SCO2 mutation in an early onset rapidly progressive, fatal cardiomyopathy. Mol Genet Metab 2006; 89: 129–133
14) Antonicka H, Leary SC, Guercin GH, et al: Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum Mol Genet 2003; 12: 2693–2702
15) Antonicka H, Mattman A, Carlson CG, et al: Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am J Hum Genet 2003; 72: 101–114
16) Diodato D, Invernizzi F, Lamantea E, et al: Common and Novel TMEM70 Mutations in a Cohort of Italian Patients with Mitochondrial Encephalocardiomyopathy. JIMD Rep 2015; 15: 71–78
17) Mazurova S, Magner M, Kucerova-Vidrova V, et al: Thymidine kinase 2 and alanyl-tRNA synthetase 2 deficiencies cause lethal mitochondrial cardiomyopathy: Case reports and review of the literature. Cardiol Young 2017; 27: 936–944
18) Gotz A, Tyynismaa H, Euro L, et al: Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet 2011; 88: 635–642
19) Takeda A, Sudo A, Yamada M, et al: Eponym: Barth syndrome. Eur J Pediatr 2011; 170: 1365–1367
20) Siriwardena K, Mackay N, Levandovskiy V, et al: Mitochondrial citrate synthase crystals: Novel finding in Sengers syndrome caused by acylglycerol kinase (AGK) mutations. Mol Genet Metab 2013; 108: 40–50
21) Strauss KA, DuBiner L, Simon M, et al: Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc Natl Acad Sci USA 2013; 110: 3453–3458
22) El-Hattab AW, Scaglia F: Mitochondrial Cardiomyopathies. Front Cardiovasc Med 2016; 3: 25
23) Marin-Garcia J, Goldenthal MJ: The mitochondrial organelle and the heart. Revista espanola de cardiologia 2002; 55: 1293–1310
24) Scaglia F, Towbin JA, Craigen WJ, et al: Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics 2004; 114: 925–931
25) Bates MG, Nesbitt V, Kirk R, et al: Mitochondrial respiratory chain disease in children undergoing cardiac transplantation: A prospective study. Int J Cardiol 2012; 155: 305–306
26) Finsterer J: Cardiogenetics, neurogenetics, and pathogenetics of left ventricular hypertrabeculation/noncompaction. Pediatr Cardiol 2009; 30: 659–681
27) Finsterer J: Histiocytoid cardiomyopathy: A mitochondrial disorder. Clin Cardiol 2008; 31: 225–227
28) Finsterer J, Kothari S: Cardiac manifestations of primary mitochondrial disorders. Int J Cardiol 2014; 177: 754–763
29) Anan R, Nakagawa M, Miyata M, et al: Cardiac involvement in mitochondrial diseases: A study on 17 patients with documented mitochondrial DNA defects. Circulation 1995; 91: 955–961
30) Majamaa-Voltti K, Peuhkurinen K, Kortelainen ML, et al: Cardiac abnormalities in patients with mitochondrial DNA mutation 3243A>G. BMC Cardiovasc Disord 2002; 2: 12
31) Catteruccia M, Sauchelli D, Della Marca G, et al: “Myo-cardiomyopathy” is commonly associated with the A8344G “MERRF” mutation. J Neurol 2015; 262: 701–710
32) Barrera-Ramirez CF, Barragan-Campos HM, Ilarraza H, et al: Cardiac involvement in Kearns-Sayre syndrome. Rev Esp Cardiol 2005; 58: 443–446
33) Bernier FP, Boneh A, Dennett X, et al: Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 2002; 59: 1406–1411
34) Ogawa E, Shimura M, Fushimi T, et al: Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: A study of 106 Japanese patients. J Inherit Metab Dis 2017; doi:10.1007/s10545-017-0042-6 [Epub ahead of print]


This page was created on 2017-07-21T09:10:05.58+09:00
This page was last modified on 2017-08-14T17:04:25.145+09:00
このサイトは(株)国際文献社によって運用されています。