Barth症候群Barth Syndrome
福島県立医科大学医学部小児科Department of Pediatrics, Fukushima Medical University ◇ Fukushima, Japan
発行日:2016年9月1日Published: September 1, 2016
バース症候群はX連鎖性の疾患であり,心筋症,好中球減少,ミオパチー,成長障害,3-メチルグルタコンサン尿症を特徴とする.本疾患はXq28に存在するTAZ遺伝子の変異によって発症し,カルジオリピンの欠損とミトコンドリア機能異常をきたす.心筋疾患は,Barth症候群において最も多く認められる症状であり,拡張型心筋症と左室心筋緻密化障害が見られ,頻度は少ないが心内膜線維弾性症や肥大型心筋症が見られる.好中球減少やミオパチーなどの症状がなく,TAZ遺伝子異常が認められるX連鎖性の乳児心筋症が報告されており,これらはBarth症候群とallelicであると考えられている.本症候群による死亡の大半は心不全によるものであり,その多くは生後6か月以内に生じる.一方,治療に対する反応は良好であり,3歳以降は心機能が正常化する症例が多く見られる.不整脈が年長児で見られることがあるが,心機能が改善した5歳以降の予後は良好であることから,本症候群の表現型の多様性を理解し,早期診断と早期治療が重要である.
Barth syndrome is an X-linked disorder characterized by cardiomyopathy, neutropenia, skeletal myopathy, growth delay, and increased urinary excretion of 3-methylglutaconic acid. The disorder is caused by mutations in the TAZ gene at Xq28 that result in cardiolipin deficiency and abnormal mitochondria. Cardiac involvement is the most common feature of this syndrome, which usually includes dilated cardiomyopathy or left ventricular noncompaction, and less commonly includes endocardial fibroelastosis or hypertrophic cardiomyopathy. X-linked infantile cardiomyopathies with mutations in the TAZ gene but without Barth syndrome-related symptoms, such as neutropenia and skeletal myopathy, have been reported and postulated to be allelic variants of Barth syndrome. Heart failure is a leading cause of death in such patients, with the highest incidence during the first 6 months of life. In contrast, many patients appear to respond to conventional medical therapy, and cardiac function usually normalizes after 3 years of age. Although Barth syndrome is associated with an increased risk of ventricular arrhythmia, the prognosis of most patients is good after 5 years of age when cardiac function subsequently recovers during infancy. Therefore, early diagnosis and management, with recognition of a wide range of phenotypes, are important for improving the prognosis of Barth syndrome.
Key words: barth syndrome; mutation; cardiomyopathy; neutropenia; myopathy
© 2016 特定非営利活動法人日本小児循環器学会© 2016 Japanese Society of Pediatric Cardiology and Cardiac Surgery
Barth症候群は,拡張型心筋症,好中球減少症,骨格筋障害(ミオパチー)を呈するX連鎖性遺伝形式の比較的まれな疾患である.1993年にPeter G. Barthが,上記3徴と病理学的にミトコンドリア形態異常を認めるX連鎖性遺伝形式の大家系を報告したことにちなんで命名された1, 2).その後,Kelleyらによって,尿中への3-メチルグルタコン酸排泄が報告され3),本疾患は3-メチルグルタコン酸尿症II型に分類された4–6).家系解析により,X染色体Xq28上のTAZ(G4.5)の遺伝子変異が原因であることが判明し7–9),現在まで多くの種類の変異が報告されている.TAZ遺伝子変異があるが,好中球減少やミオパチー等のBarth症候群の症状を認めない心筋症の症例があり10–14),これらはBarth症候群とallelicな類縁疾患である.
人種差や地域差はなく,30~40万人に1人の発生頻度とされるが15),早期死亡例や軽症例など,診断されていない症例の存在が示唆されている16, 17),心筋症以外の症状がなくTAZ遺伝子変異を認める例を類縁疾患と考えBarth症候群に加えると,その発生頻度は更に高くなる,疾病および関連保健問題の国際統計分類(ICD-10)の2015年改訂において,Barth症候群がE78.71としてコード化されたため,今後,詳細な疫学がわかってくることが期待される,日本でも数家系の報告がある18–23).
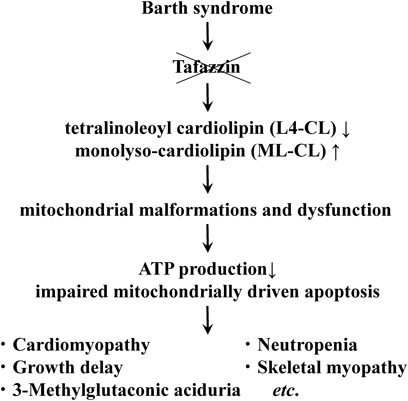
Xq28上のTAZ(G4.5)が責任遺伝子である7–9),TAZ(G4.5)がコードする蛋白はTafazzinで,アシルトランスフェラーゼに属すると考えられている24),アシルトランスフェラーゼは,ミトコンドリア膜に存在し,カルジオリピン(CL)やホスファチジルグリセオールなどのリン脂質の合成に関与し,Barth症候群ではミトコンドリア内膜に存在するCLの含量が低下する25, 26),CLは四つの脂肪酸鎖を有するリン脂質で,正常の心筋・骨格筋ではCLの約80%が四つのリノール酸を有するL4-CL(tetralinoleoyl cardiolipin)であるが,Barth症候群ではL4-CLが減少し,その前駆物質のML-CL(monolyso-cardiolipin)が蓄積する,このCL含量低下と組成の変化が,ミトコンドリアの構造や機能,アポトーシスに異常をきたし,ATPの産生能力が低下するために,心筋症や骨格筋障害等の症状が出現すると推測されている25, 27–30)(Fig. 1).
TAZ遺伝子には11個のコドンが存在し,これまでに200種類以上のTAZ遺伝子変異が報告され5, 31),Barth syndrome foundationのホームページ(https://www.barthsyndrome.org/home)にHuman TAZ Gene Variants Databaseが公開されている,TAZ遺伝子変異の部位・種類と,表現型・重症度との関連は証明されておらず31),家系内で重症度の異なる症例も報告されている32).しかし,後述のように,本疾患は新生児期・乳児期に死亡が多い反面,その後は症状が軽減していく特徴を有し,早期発見と早期介入により予後が大幅に改善されることから,早期診断例が蓄積されていくと遺伝子型と表現型の関連性が判明してくる可能性もある,母親が保因者の場合,男児の50%に本疾患が発症する,母親に遺伝子変異が見られず,児に新規の変異が認められるのは20%以下であるが33),我々は母親に遺伝子変異がない同胞例を経験し,性腺モザイク例として報告した22, 23).
本症候群は,拡張型心筋症,好中球減少症,ミオパチーを3徴とするが,そのほか,Table 1のような症状が報告されている15, 33).RobertsらはBarth syndrome foundationの患者レジストリに登録された73例をまとめ,症状発現年齢は0.76±1.6歳で,本疾患と確定診断された年齢は4.04±5.45歳と報告しているが,本疾患の理解が進めば,診断年齢は低下するものと思われる.
| Cardiovascular | Left ventricular noncompaction, dilated cardiomyopathy, endocardial fibroelastosis, ventricular arrhythmias, sudden cardiac death |
| Hematologic | Neutropenia |
| Infectious | Recurrent bacterial infections, recurrent oral ulcers and sore gums, perianal dermatitis, diarrhea |
| Skeletal muscular | Proximal myopathy, easy fatigability, exercise intolerance |
| Endocrine & Metabolic | 3-methylglutaconic aciduria, growth delay, delayed bone age, osteopenia, hypocholesterolemia, high ML-CL/L4-CL ratio |
| Dysmorphic | Full cheeks, deep set eyes, prominent ears |
| Developmental | Delayed motor milestones, learning disabilities, attention deficit disorder, oromotor feeding abnormalities |
| Genetic | TAZ (G4.5) gene mutations |
| Fetal | Cardiomyopathy, fetal hydrops, male miscarriage, and stillbirth |
心筋症はBarth症候群で最も多く見られる症候で,Barth syndrome foundationの患者レジストリでは73人中69人に心疾患を認め,うち48人(74%)が1歳までに心筋症の診断を受けていた34).拡張型心筋症および左室心筋緻密化障害が主たる表現型であるが,心内膜線維弾性症8, 35)を合併することもある.心筋肥厚の報告もあるが,左室心筋緻密化障害との関連が示唆されている36).オーストラリアで1987年から1998年までの10年間に新規に診断された10歳未満の心筋症は314人(10万対罹患率1.24)で,男性166人中8人(4.8%)がBarth症候群であった37).心筋症の男児をみた場合には,Barth症候群を考慮することが必要である.胎児期の心筋症発症もあり,胎児診断例35, 38)や剖検例39)が報告されている.英国のBarth症候群6家系の調査で,男性において14人の小児期死亡と9人の死産があるのに対し,女性では死産や小児期死亡がなく,また同家系内に18人の流産が認められた.これらの死産・流産はBarth症候群の胎児期発症例と考えられる40).
心筋症の予後については,早期死亡が見られる一方で,成長とともに心機能が改善していくことが報告されている.Kangらは,英国の27人のBarth症候群患者について,臨床症状と心臓超音波所見の推移を報告した41).これによると,27人中25人が診断当初より心筋症を有しており,22人は生後6か月以内に診断されていた.27人中,22人が生存し,5人が0.02~4.22歳(1.8歳)で心不全に起因して死亡した.生存例の2.0~22歳(中央値12.6歳)までの心臓超音波検査による経過観察では,心筋ストレイン解析において異常が残存するものの,多くの症例で心機能は3歳以降に改善し,拡張末期径,左室内径短縮率,左室拡張末期圧指標E/E’は正常化した.フランスにおける22症例の研究では,22人中11人が死亡していたが,2000年以前の出生では生存率が20%であったのに対し,2000年以降の出生では生存率が80%に改善し,心機能は同様に3歳以降改善していくことが示された15, 42).Fig. 2に自験例の治療経過を提示した.2か月時に哺乳力低下と体重増加不良を契機に発見され,入院時,胸部X線では心胸郭比65%の心拡大と軽度の肺うっ血を認め(Fig. 2A),BNP値は725 pg/mLと上昇し,心臓超音波検査では左室心筋緻密化障害の所見と心拡大,収縮力低下,中等度の僧帽弁閉鎖不全を認めた(Fig. 2B).βブロッカー,アンジオテンシン変換酵素阻害薬,利尿薬による治療により,1歳過ぎには心拡大は消失し(Fig. 2C)BNPは正常化した.7歳時においても心機能は良好に保たれ,心臓超音波検査では左室心筋緻密化障害の所見と少量の僧帽弁閉鎖不全は認めるものの,心収縮力は良好に保たれている(Fig. 2D).本疾患における心筋障害は,呼吸器感染症などを契機に発見されることもあり,このような心筋障害の回復に加え,好中球減少が感染による骨髄抑制と考えられてしまうことで,ウイルス性心筋炎/心筋症と誤診されることが示唆されている15).

Chest roentgenogram at the age of 2 months revealing cardiomegaly with a cardiac ratio of 65% and mild pulmonary edema (Fig. 2A). Echocardiogram demonstrating left ventricular noncompaction, left ventricular dilation with LVIDD of 29 mm, poor contractility with LVFS of 13%, and moderate mitral regurgitation (Fig. 2B; movie). Chest roentgenogram at the age of 13 months revealing a normal heart size (Fig. 2C). Echocardiogram at the age of 7 years revealing normal cardiac function with LVIDD of 37 mm, LVFS of 40%, and mild mitral regurgitation (Fig. 2D; movie).
心電図異常も多くの症例で見られ,主たる異常は,左軸偏位,左室肥大,脚ブロック,ST-T異常,QT延長などである41, 43).QT延長についてKangらは43%が460 msecを超え,14%が450~460 msecと報告し41),Spencerらは20%が460 msec以上で,23%が450~459 msecと報告している44).治療を要する心室性不整脈の頻度は,報告によって大きく異なる.Kangらの27例の報告では,重症な拡張型心筋症を合併していた新生児1例のみに心室性不整脈を認めたとしているが,Spencerらは34人中7人(20.5%)に心室性不整脈を認め,死亡した1例を除き植え込み型除細動器(ICD)の治療を受け,Barth syndrome foundationによる患者レジストリでも,12.9%の患者がICD治療を受けていた.心機能低下の程度と不整脈のリスクには関連が示されておらず15, 45),QTc時間も不整脈リスクと無関係とされている43, 45).
TAZ遺伝子変異を認めながら,好中球減少やミオパチー等のBarth症候群に見られる症状を認めない拡張型心筋症,左室心筋緻密化障害や心筋肥厚を伴う心内膜線維弾性症などの症例や家系が報告されており10–14),Barth症候群とallelicであることが示されている.
好中球減少症は,心筋症についで多く見られる症状であるが,個人差があり,経時的変動が見られ,正常化することもある3, 15).Barth syndrome foundationによる患者レジストリでは,69.1%に好中球減少が見られた34).Barthらは,前骨髄球の増加と骨髄球の減少を認め,骨髄球の成熟障害を示唆しているが1),最近は,機序としてマクロファージによる好中球クリアランスの亢進が推測されている46, 47).好中球の遊走能と殺菌能は正常に保たれる48).初期の報告では,敗血症などの重篤な感染症により心不全と同等の死亡が見られたが1),現在は,感染症単独での死亡例は減少した.しかし,反復する口腔内潰瘍や歯肉炎などが好中球減少に関連し49),肺炎や血液感染症の罹患率も高い34).
骨格筋障害として近位筋を中心に軽度から中等度の筋力低下が見られ,乳幼児期の粗大運動発達遅滞が見られる1, 15, 43).しかし,ほとんどの児は2歳までに補助なしで歩行することができるようになり2),筋力低下は10歳までの間,成長とともに軽快し,その後安定する43).思春期以降は,日常生活に支障が出ることはまれであるが,易疲労を訴えることが多い.運動負荷試験を行うと,ミトコンドリア機能に関連する骨格筋の酸素摂取・利用に障害を認め,加えて心臓予備能の低さから,コントロール群に比して運動耐容能の低下がある44).筋生検では,タイプ2線維の萎縮,タイプ1線維内の脂肪滴沈着やチトクロームc酸化酵素染色のびまん性低下などを認める27, 50).
本疾患で認められる3-メチルグルタコン酸尿症は,ミトコンドリア機能障害による二次的な3-メチルグルタコン酸増加が原因と考えられるが,その機序は十分に解明されていない27).現在,3-メチルグルタコンサン尿症は5型に分けられ,Barth症候群はII群に分類されている5, 6).3-メチルグルタコン酸尿は診断に有用であるが,TAZ遺伝子変異がありながら尿中排泄が見られない症例も報告されている10, 11, 39, 50).また,日内変動があり,ほかの3-メチルグルタコンサン尿症に比して排泄量も少ないことから16),診断に用いる際は注意が必要である.
そのほかの代謝異常としては,LDL(low density lipoprotein)コレステロールやプレアルブミンの低下が報告されている43).また早期死亡例で,代謝性アシドーシス,低血糖,トランスアミナーゼ高値,高乳酸血症,高アンモニア血症などが報告されている51, 52).ミトコンドリア機能異常が代謝障害をきたし早期死亡に関与することも推測されたが,重症心不全に伴う二次的な異常とも考えられる.また,代謝異常を示さず死亡する症例もあり,ミトコンドリア機能異常と早期死亡の関連については議論がある.
発育障害は多くの症例で見られ,思春期前は−3SD~−2SDの低身長を認め,骨年齢は遅れる15, 26).しかし,12~14歳以降,身長の伸びは良好となり,通常,身長の伸びが止まってくる思春期以降も伸び続け,成人で正常となる例も多い15, 34).Wilsonらの検討では,Barth症候群患児の成長ホルモンは,14.4歳未満ではコントロール群に比較し低値であったが,14.4歳以降はコントロール群を上回っていた53).本疾患で血清アルギニン値が低値であることが示され,そのためにタンパク質合成が阻害され,成長に悪影響を与えることが報告されている42).
50~70%の乳幼児に,多くは生後6か月以前から出現する経口摂取困難が見られ,発育障害の一因と考えられている54).嘔吐しやすく,咀嚼力が弱く,少量ずつの摂取で食事に時間がかかり,離乳が遅れる15).Barth syndrome foundationによる患者レジストリでは,約1/3の症例が,この時期に経管栄養を要した34).最近,Barth症候群では味覚認識が異なっており,塩辛い食品を好むことが経口摂食の進まない一因であると報告されている54, 55).
成人では目立たなくなるが,小児期の顔貌には特徴がある.幼少期はやや突出した幅広い額に,丸顔でふっくらとした頬をしており,目はやや落ち窪んで,耳介は大きめで立っている.思春期になると丸顔は細くなり,特徴がなくなってくる15, 56).
認知能力に関しては,5歳から10歳のBarth症候群児を対象とした研究で,読解に関連した能力は正常であるが,数学や視覚的・空間的認識や視覚的な短期記憶が,同年齢の児に劣ると報告された57).普通学校に通学が可能な例が多いと考えられるが,作業療法や言語療法を要する場合も多く34),児の状況に合わせた学習支援の計画が必要である.
拡張型心筋症または左室心筋緻密化障害,好中球減少症,ミオパチーおよび3-メチルグルタコン酸尿症が古典的な症状であるが,TAZ遺伝子変異を有しながら,症状が揃わない症例があることに注意を要する.好中球減少は経時的に変化し正常であることも多く,ミオパチーも軽微であり症状を呈さないこともある.検査所見として3-メチルグルタコン尿症は診断に有用であるが,正常例もある.心筋症は最も頻度が高い症状であり,収縮能が低下していなくても左室心筋緻密化障害の所見が見られる男児に対しては,本疾患を疑う.死産や出生後早期死亡の家族歴を有したり,発育・発達障害やミオパチーが疑われたりする男児に対しては,心臓超音波検査と尿中有機酸分析を行うとともにTAZ遺伝子変異を調べることが確定診断につながる.
海外の幾つかの施設では,濾紙血で検査可能なタンデムマス分析によるML-CL/L4-CL比によるスクリーニングが行われている58).また,近年,白血球を検体として液体クロマトグラフ-タンデム型質量分析計(LC-MS/MS)によるML-CL/L4-CL比測定の有用性が報告されている.Barth症候群ではML-CL/L4-CL比は高値を示すが,LC-MS/MSの測定では,Barth症候群症例の最低値がコントロール群の最高値の400倍以上にあたり,100%の感度と特異度をもって迅速に診断可能である59).
胎児診断については,1997年に初めて胎児期に診断された症例が報告されたが,在胎21週では異常がなく,在胎33.5週で発育障害と心拡大で発見された38).在胎18週の胎児の剖検例の心臓において,明らかな心肥大と心内膜線維弾性症所見が見られるという報告がある一方39),24~30週の胎児では異常が見られない場合もあり,超音波検査による出生前診断には限界がある23, 35).
心筋症に関しては,心機能の改善が見られている多くの症例で,アンジオテンシン変換酵素阻害薬やβブロッカーが投与されている41).Barth症候群の心筋症に特異的に有効な薬剤は証明されておらず,一般的な心不全療法が有効であると考えられている15).βブロッカーとしては,carvedilol, metoprolol, bisoprololが多く使用されているが,有用性の差に関する報告はない.新生児から乳児期に高度の心不全に陥り,さらなる心筋障害をきたすことのないように,早期診断と早期の治療開始が重要である23, 41, 58).海外では重症例に対する心臓移植も行われ,Barth syndrome foundationによる患者レジストリでは12.3%の症例に行われていた34).英国からの報告では,22人中7人に心臓移植が行われ,生存した6人は無症状で左室機能も良好に保たれている.重症例に対する心臓移植は有用であると考えられるが,心機能は徐々に改善していくことを念頭に入れて適応を考える必要があり2),今後,早期の内科的治療が行われるようになれば,適応患者は減少することが期待される.不整脈に関しては,重篤な不整脈が10歳以上の年長児にも見られているため45),心機能が改善してきても安心することなく観察することが重要である.治療としては,抗不整脈薬の効果に関する報告はなく,先述のような植え込み型除細動器(ICD)による治療が報告されている.
好中球減少に対する治療としては,顆粒球コロニー刺激因子(GCSF)の投与や抗菌薬投与が行われる15).好中球減少は経時的変動が大きいため,GCSF投与は正常値までの増加を目指す必要はなく,投与により,アフタ性潰瘍や歯肉炎の予防,細菌性感染の減少,時には倦怠感の改善が得られる15).ミオパチーは,成人までには症状が改善し,日常生活に支障をきたすことはまれである.しかし,運動耐容能は正常より低いため,心不全の状態と合わせて,運動制限を考える必要がある.低身長は思春期以降に改善が認められるため,成長ホルモンの投与は通常行われない.カルニチン投与60)やパントテン酸投与61)の著効した症例が報告されているが,有用性が証明されておらず,逆に無効例や投与によって悪化した症例も報告されており61, 62),一般に推奨されない15, 49).アルギニン投与が発育障害の改善に有用であることが示唆されたが,証明されてはいない42).
Barth症候群は,心疾患で発症することが多いため,小児循環器医師が関わることが多いと考えられる.早期治療介入の重要性と表現型の多様性に留意して診療にあたることが望まれる.
本論文について,開示すべき利益相反(COI)はない.
この論文の電子版にて動画を配信している.
1) Barth PG, Scholte HR, Berden JA, et al: An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci 1983; 62: 327–355
2) Barth PG, Valianpour F, Bowen VM, et al: X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): An update. Am J Med Genet A 2004; 126A: 349–354
3) Kelley RI, Cheatham JP, Clark BJ, et al: X-linked dilated cardiomyopathy with neutropenia, growth retardation, and 3-methylglutaconic aciduria. J Pediatr 1991; 119: 738–747
4) Gibson KM, Sherwood WG, Hoffman GF, et al: Phenotypic heterogeneity in the syndromes of 3-methylglutaconic aciduria. J Pediatr 1991; 118: 885–890
5) 坂本 修:有機酸・脂肪酸代謝異常分枝有機酸尿症3-メチルグルタコン酸尿症II型(Barth症候群).日本臨床2012; 別冊 先天代謝異常症候群(第2版)(上)—病因・病態研究,診断・治療の進歩—,pp 375–377
6) 坂本 修:有機酸・脂肪酸代謝異常分枝有機酸尿症3-メチルグルタコン酸尿症I型(3-メチルグルタコニルCoAヒドラターゼ欠損症).日本臨床2012; 別冊 先天代謝異常症候群(第2版)(上)—病因・病態研究,診断・治療の進歩—,pp 372–374
7) Bolhuis PA, Hensels GW, Hulsebos TJ, et al: Mapping of the locus for X-linked cardioskeletal myopathy with neutropenia and abnormal mitochondria (Barth syndrome) to Xq28. Am J Hum Genet 1991; 48: 481–485
8) Adès LC, Gedeon AK, Wilson MJ, et al: Barth syndrome: Clinical features and confirmation of gene localisation to distal Xq28. Am J Med Genet 1993; 45: 327–334
9) Bione S, D’Adamo P, Maestrini E, et al: A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet 1996; 12: 385–389
10) Gedeon AK, Wilson MJ, Colley AC, et al: X linked fatal infantile cardiomyopathy maps to Xq28 and is possibly allelic to Barth syndrome. J Med Genet 1995; 32: 383–388
11) Bleyl SB, Mumford BR, Thompson V, et al: Neonatal, lethal noncompaction of the left ventricular myocardium is allelic with Barth syndrome. Am J Hum Genet 1997; 61: 868–872
12) Chen R, Tsuji T, Ichida F, et al: Noncompaction study collaborators: Mutation analysis of the G4.5 gene in patients with isolated left ventricular noncompaction. Mol Genet Metab 2002; 77: 319–325
13) Ichida F, Tsubata S, Bowles KR, et al: Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 2001; 103: 1256–1263
14) D’Adamo P, Fassone L, Gedeon A, et al: The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am J Hum Genet 1997; 61: 862–867
15) Clarke SLN, Bowron A, Gonzalez IL, et al: Barth syndrome. Orphanet J Rare Dis 2013; 8: 23
16) Cantlay AM, Shokrollahi K, Allen JT, et al: Genetic analysis of the G4.5 gene in families with suspected Barth syndrome. J Pediatr 1999; 135: 311–315
17) 市田蕗子:特集 知っておきたい内科症候群II.循環器《先天性疾患》バース症候群.内科2012; 109: 1037–1038
18) Sakamoto O, Ohura T, Katsushima Y, et al: A novel intronic mutation of the TAZ (G4.5) gene in a patient with Barth syndrome: Creation of a 5′ splice donor site with variant GC consensus and elongation of the upstream exon. Hum Genet 2001; 109: 559–563
19) Sakamoto O, Kitoh T, Ohura T, et al: Novel missense mutation (R94S) in the TAZ (G4.5) gene in a Japanese patient with Barth syndrome. J Hum Genet 2002; 47: 229–231
20) Katsushima Y, Fujiwara I, Sakamoto O, et al: Normal pituitary function in a Japanese patient with Barth syndrome. Eur J Pediatr 2002; 161: 67–68
21) Xing Y, Ichida F, Matsuoka T, et al: Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol Genet Metab 2006; 88: 71–77
22) Chang B, Momoi N, Shan L, et al: Noncompaction study collaborators: Gonadal mosaicism of a TAZ (G4.5) mutation in a Japanese family with Barth syndrome and left ventricular noncompaction. Mol Genet Metab 2010; 100: 198–203
23) Momoi N, Chang B, Takeda I, et al: Differing clinical courses and outcomes in two siblings with Barth syndrome and left ventricular noncompaction. Eur J Pediatr 2011; 171: 515–520
24) Neuwald AF: Barth syndrome may be due to an acyltransferase deficiency. Curr Biol 1997; 7: R465–R466
25) Vreken P, Valianpour F, Nijtmans LG, et al: Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun 2000; 279: 378–382
26) 坂本 修,大浦敏博,飯沼一宇:小児医学最近の進歩Barth症候群の分子遺伝学と臨床.小児科2002; 43: 2011–2016
27) 武田充人,須藤 章:先天代謝異常によるミオパチー先天性筋脂質代謝異常症中性脂質代謝異常症Barth症候群.日本臨床2015; 別冊:130–134
28) Schlame M, Kelley RI, Feigenbaum A, et al: Phospholipid abnormalities in children with Barth syndrome. J Am Coll Cardiol 2003; 42: 1994–1999
29) Gonzalvez F, Gottlieb E: Cardiolipin: Setting the beat of apoptosis. Apoptosis 2007; 12: 877–885
30) Saric A, Andreau K, Armand A-S, et al: Barth Syndrome: From Mitochondrial Dysfunctions Associated with Aberrant Production of Reactive Oxygen Species to Pluripotent Stem Cell Studies. Front Genet 2015; 6: 359
31) Johnston J, Kelley RI, Feigenbaum A, et al: Mutation characterization and genotype-phenotype correlation in Barth syndrome. Am J Hum Genet 1997; 61: 1053–1058
32) Ronvelia D, Greenwood J, Platt J, et al: Intrafamilial variability for novel TAZ gene mutation: Barth syndrome with dilated cardiomyopathy and heart failure in an infant and left ventricular noncompaction in his great-uncle. Mol Genet Metab 2012; 107: 428–432
33) Jefferies JL: Barth syndrome. Am J Med Genet C Semin Med Genet 2013; 163C: 198–205
34) Roberts AE, Nixon C, Steward CG, et al: The Barth Syndrome Registry: Distinguishing disease characteristics and growth data from a longitudinal study. Am J Med Genet A 2012; 158A: 2726–2732
35) Bleyl SB, Mumford BR, Brown-Harrison MC, et al: Xq28-linked noncompaction of the left ventricular myocardium: Prenatal diagnosis and pathologic analysis of affected individuals. Am J Med Genet 1997; 72: 257–265
36) Pignatelli RH, McMahon CJ, Dreyer WJ, et al: Clinical characterization of left ventricular noncompaction in children: A relatively common form of cardiomyopathy. Circulation 2003; 108: 2672–2678
37) Nugent AW, Daubeney PEF, Chondros P, et al: National Australian Childhood Cardiomyopathy Study: The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med 2003; 348: 1639–1646
38) Cardonick EH, Kuhlman K, Ganz E, et al: Prenatal clinical expression of 3-methylglutaconic aciduria: Barth syndrome. Prenat Diagn 1997; 17: 983–988
39) Brady AN, Shehata BM, Fernhoff PM: X-linked fetal cardiomyopathy caused by a novel mutation in the TAZ gene. Prenat Diagn 2006; 26: 462–465
40) Steward CG, Newbury-Ecob RA, Hastings R, et al: Barth syndrome: An X-linked cause of fetal cardiomyopathy and stillbirth. Prenat Diagn 2010; 30: 970–976
41) Kang S-L, Forsey J, Dudley D, et al: Clinical Characteristics and Outcomes of Cardiomyopathy in Barth Syndrome: The UK Experience. Pediatr Cardiol 2015; 37: 167–176
42) Rigaud C, Lebre AS, Touraine R, et al: Natural history of Barth syndrome: A national cohort study of 22 patients. Orphanet J Rare Dis 2013; 8: 70
43) Spencer CT, Bryant RM, Day J, et al: Cardiac and clinical phenotype in Barth syndrome. Pediatrics 2006; 118: e337–e346
44) Spencer CT, Byrne BJ, Bryant RM, et al: Impaired cardiac reserve and severely diminished skeletal muscle O2 utilization mediate exercise intolerance in Barth syndrome. Am J Physiol Heart Circ Physiol 2011; 301: H2122–H2129
45) Spencer CT, Byrne BJ, Gewitz MH, et al: Ventricular arrhythmia in the X-linked cardiomyopathy Barth syndrome. Pediatr Cardiol 2005; 26: 632–637
46) Finsterer J, Frank M: Haematological features in Barth syndrome. Curr Opin Hematol 2013; 20: 36–40
47) van Raam BJ, Kuijpers TW: Mitochondrial defects lie at the basis of neutropenia in Barth syndrome. Curr Opin Hematol 2009; 16: 14–19
48) Kuijpers TW, Maianski NA, Tool Anton TJ, et al: Neutrophils in Barth syndrome (BTHS) avidly bind annexin-V in the absence of apoptosis. Blood 2004; 103: 3915–3923
49) Huhta JC, Pomerance HH, Barness EG: Clinicopathologic conference: Barth Syndrome. Fetal Pediatr Pathol 2005; 24: 239–254
50) Takeda A, Sudo A, Yamada M, et al: Barth syndrome diagnosed in the subclinical stage of heart failure based on the presence of lipid storage myopathy and isolated noncompaction of the ventricular myocardium. Eur J Pediatr 2011; 170: 1481
51) Yen T-Y, Hwu W-L, Chien Y-H, et al: Acute metabolic decompensation and sudden death in Barth syndrome: report of a family and a literature review. Eur J Pediatr 2008; 167: 941–944
52) Donati MA, Malvagia S, Pasquini E, et al: Barth syndrome presenting with acute metabolic decompensation in the neonatal period. J Inherit Metab Dis 2006; 29: 684
53) Wilson Lori D, Al-Majid S, Rakovski CS, et al: Higher IL-6 and IL6:IGF Ratio in Patients with Barth Syndrome. J Inflamm (Lond) 2012; 9: 25
54) Reynolds S, Kreider CM, Meeley LE, et al: Taste perception and sensory sensitivity: Relationship to feeding problems in boys with Barth Syndrome. J Rare Disord 2015; 3: 1–9
55) Reynolds S, Kreider CM, Bendixen R: A mixed-methods investigation of sensory response patterns in Barth syndrome: A clinical phenotype? Am J Med Genet A 2012; 158A: 1647–1653
56) Hastings R, Steward C, Tsai-Goodman B, et al: Dysmorphology of Barth syndrome. Clin Dysmorphol 2009; 18: 185–187
57) Mazzocco MMM, Henry AE, Kelly RI: Barth syndrome is associated with a cognitive phenotype. J Dev Behav Pediatr 2007; 28: 22–30
58) Kulik W, van Lenthe H, Stet FS, et al: Bloodspot assay using HPLC-tandem mass spectrometry for detection of Barth syndrome. Clin Chem 2008; 54: 371–378
59) Bowron A, Frost R, Powers VEC, et al: Diagnosis of Barth syndrome using a novel LC-MS/MS method for leukocyte cardiolipin analysis. J Inherit Metab Dis 2013; 36: 741–746
60) Ino T, Sherwood WG, Cutz E, et al: Dilated cardiomyopathy with neutropenia, short stature, and abnormal carnitine metabolism. J Pediatr 1988; 113: 511–514
61) Ostman-Smith I, Brown G, Johnson A, et al: Dilated cardiomyopathy due to type II X-linked 3-methylglutaconic aciduria: Successful treatment with pantothenic acid. Br Heart J 1994; 72: 349–353
62) Rugolotto S, Prioli MD, Toniolo D, et al: Long-term treatment of Barth syndrome with pantothenic acid: A retrospective study. Mol Genet Metab 2003; 80: 408–411


This page was created on 2016-08-30T15:06:49.584+09:00
This page was last modified on 2016-09-28T20:09:59.72+09:00
このサイトは(株)国際文献社によって運用されています。