肺高血圧症(pulmonary hypertension: PH)の遺伝学的背景を考える上で,まずは2018年に開催された6th World Symposium on Pulmonary Hypertension(WSPH)で定められた,肺動脈性肺高血圧症(pulmonary arterial hypertension: PAH)の原因遺伝子を押さえておく必要がある.6th WSPHにおいては,それぞれのエビデンスレベルに基づき,BMPR2をはじめとした17遺伝子が疾患原因遺伝子として挙げられた(Table 1)1).このうちSMAD9(SMAD8),BMPR1Bの2遺伝子は,東京女子医科大学において世界で初めて同定されたものである2, 3).17遺伝子のうちのひとつ,SOX17についてはその希少なバリアントが,先天性心疾患に伴うPAH(congenital heart disease-associated PAH)の約3%で検出されたとの報告が2018年になされており,二次性PAHの原因を考える上で非常に興味深い4).
Table 1 Classification of pulmonary arterial hypertension genes according to level of evidence that they play a causal role in the disease [Adapted from Ref. 1)]| Higher level of evidence | Lower level of evidence |
|---|
| BMPR2; EIF2AK4; TBX4; ATP13A3; GDF2; SOX17; AQP1; ACVRL1; SMAD9; ENG; KCNK3; CAV1 | SMAD4; SMAD1; KLF2; BMPR1B; KCNA5 |
| Evidence includes de novo mutation, cosegregation studies, association with replication and functional studies. |
さらにその後も,新たな疾患原因遺伝子候補についての報告が相次いでいる.前回の総説5)でも紹介したが,2018年にはATP依存性カリウムチャネルであり,高インスリン血症の原因遺伝子としても知られているABCC8遺伝子の病的バリアントが12名のPAH患者で同定されている6).そして,高インスリン血症の治療薬であるジアゾキサイドがこの変異カリウムチャネルの機能を改善させたと報告されている.
また,2020年には,特発性PAH(idiopathic PAH: IPAH)230名において全ゲノム解析を行い,14名において計3種類のPTGIS遺伝子の希少なバリアントを同定したとの報告が中華人民共和国からなされている7).このPTGIS遺伝子はプロスタサイクリン合成酵素をコードしている.PTGIS希少なバリアントを有していた患者は,これを持たない患者と比較して,イロプロスト吸入後に肺血管抵抗がより有意に低下し,心係数も有意に上昇したことが報告されている(Fig. 1).
筆者らも,2名のPAH患者が存在する家系において全エクソーム解析を行った結果,BRAP遺伝子の病的バリアント(NM_006768.4, c.1661 G>T p.R554L)を同定して2019年に報告している.ヒト肺動脈平滑筋細胞を用いた機能解析を元に,このp.R554Lによってp53伝達経路の活性低下が惹起され,これによりヒト肺動脈平滑筋細胞が異常増殖し,その結果肺動脈肥厚が生じてPAHが発症する可能性を示した(Fig. 2)8).
PAHとp53伝達経路の関係については,これまでにも検討がなされてきた.Nutlin-3a(MDM2[p53分解を促進する制御因子]アンタゴニスト)によるp53の活性化は,ヒト肺動脈平滑筋における細胞増殖の抑制,さらには低酸素誘発性肺高血圧モデルマウスおよびSU5416/低酸素症肺高血圧モデルマウスにおける肺高血圧の予防・改善をもたらすとの報告がある9).さらに2019年には,PAHモデル動物の肺動脈平滑筋細胞においてp53が減少していること,それが低酸素誘導因子(hypoxia inducible factor-1α: HIF-1α)の増加と関連していると報告された10).
BRAP(BRCA1 associated protein)は近年,その発現が大腸癌患者の予後と関連していることや11),食道扁平上皮癌の浸潤性と関連していることが注目されている12).また,BRAPの一塩基多型(single-nucleotide polymorphism: SNP)が冠動脈疾患,心筋梗塞,高血圧,メタボリックシンドロームなどの遺伝的危険因子であることがいくつかの研究で明らかになっている13–17).
筆者らの研究で同定したBRAP p.R554Lは,既報のSNPとは全く異なる部位に位置している.その病的意義についてはさらなる検討が必要だが,BRAPが複数の悪性腫瘍の重症度との関連がある遺伝子であることは,PAHを肺動脈平滑筋細胞が異常増殖する腫瘍様疾患とみなす立場からも18, 19)エキサイティングな話題と言えるだろう.PAHの新たな治療戦略としてのp53伝達経路に,今後も注目していきたい.
修飾遺伝子に関する検討も進んでいる.PAHの原因遺伝子として最多であるBMPR2遺伝子の病的バリアントを有していたとしても,実際にPAHを発症する確率(浸透率)は平均で約20%と低い.この浸透率は男性で14%,女性で42%であることから,性差が関連していることは確実であるが20),まだ発症メカニズムには不明な点が多い.そのなかでProssedaらが,siRNAハイスループットスクリーニングを用いて,BMPR2の修飾遺伝子としてFHIT(fragile histidine triad)遺伝子を同定し,2019年に報告した.Prossedaらはさらに,プロテインキナーゼCβ選択的阻害薬であるエンザスタウリンがPAHモデルラットにおいてFHITの発現量を回復させ,さらには右室収縮期圧や非筋性肺小動脈の筋性化を改善させることを報告している21).
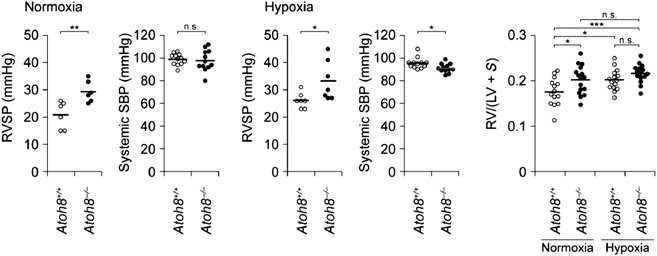
本邦からは,Morikawaらが転写因子ATOH8とPAHの関係について報告している.このグループはATOH8ノックアウトマウスがPAH同様の右室圧および肺動脈圧の上昇をきたすこと,PAH患者の肺組織ではATOH8タンパク発現量が低下していることを示し,BMPシグナル伝達経路の標的のひとつであるATOH8がPAHの発症メカニズムに関与している可能性を示唆した(Fig. 3)22).
なお,BMPR2遺伝子病的バリアントの浸透率の低さの理由として,肺高血圧症患者におけるepigeneticsにも関心は集まっている.LiuらはBMPR2変異を有する遺伝性PAH患者と,その家系内の変異キャリア(非発症者)の末梢血単核細胞を用いてBMPR2プロモーターのDNAメチル化を解析し,患者群ではそのメチル化が亢進していることを示した23).これにより,環境要因によってBMPR2の野生型対立遺伝子のメチル化が促進され,機能喪失型突然変異の浸透率が変化する可能性が示唆された.ただし,他の症例対照研究では,PAH患者と対照群の間でDNAメチル化についての有意な差は認められていない24, 25).まだまだ追加検討が必要な領域と考えられる.
2019年,Rhodesらはヨーロッパ系の健常者とPAH患者を対象とした4つの国際的な症例対象研究を用いてメタアナリシスを行い,前述の17遺伝子のひとつであるSOX17の他,HLA-DPA1/DPB1(NC_000006.11: g.33041734 T>C rs2856830)をPAH関連遺伝子として同定したと報告した26).驚くべきことに,この部位でC/Cアレルを有するPAH患者群における診断からの生存期間中央値は13.5年であり,T/Tアレルを有する群の生存期間中央値(6.97年)の約2倍であった(Fig. 4).このrs2856830はRhodesらの検討では健常ヨーロッパ人の12~13%に検出され,Japanese Multi Omics Reference Panel(jMorp: ToMMo 4.7KJPN)によると健常日本人の30%弱で検出される.今後,その意義についてより詳細な解析が待たれる.
本邦からの報告では,もやもや病の感受性遺伝子(原因遺伝子ではないことに注意)として知られているRNF213遺伝子27)に関するものも非常に興味深い.Fukushimaらが肺高血圧症ともやもや病の併発例2症例において,そのホモ接合バリアントを検出したと2016年に報告した28).その後,Kobayashiらが血管内皮細胞に特異的にRNF213変異体を発現させたトランスジェニックマウスを低酸素環境下で飼育すると,有意な右室圧上昇がみられることを2018年に報告している29).また,Suzukiらは高齢の健常者群では,PAHの原因となりうる病的バリアントは検出されないのではとの仮説を元に検討し,同じくRNF213をその候補として見いだした.RNF213希少なバリアント(NM_001256071.3: c.14429G>A p.R4810K[rs112735431])は日本人健常者の0.77%で検出されるのに対して,PAH患者群では9.2%でこのheterogenous variantが検出されたとのことであった30).さらに同じグループのHiraideは,RNF213 c.14429G>A p. R4810Kを有するPAH患者群が,BMPR2遺伝子変異を有するPAH患者群よりも有意に予後が不良とする報告を行っている31).
もやもや病の中でもこのRNF213 c.14429G>A p.R4810Kが非常に特異的なバリアントとされており,日本人のもやもや病発症者の約70%において,このバリアントが検出されることが判明している32)(注:この論文中のRNF213 c.14576 G>A p.R4859K[NM_020914.4]と,NM_001256071.3: c.14429G>A p.R4810Kは同一バリアントである).健常者集団においては,1000 Genomes Project(phase3 release V3+)によると,東アジア人で0.2%,南アジア人で0.4%検出され,アフリカ人,ヨーロッパ人,アメリカ人では検出されなかったとのことであり,人種特異性があるバリアントと考えられる.Human Genetic Variation Database(version 2.3)によると,日本人一般集団の約1%がこのバリアントを有している.そのほとんどはもやもや病を発症しないことから,このバリアントを発症原因と断定することはできない.一方で,前述のMiyawakiらはこのRNF213 c.14429G>A p.R4810Kは頭蓋内主幹動脈狭窄患者の23%で検出されたとも報告している32).さらに,前述のFukushimaの報告のうち1例は,腎動脈狭窄を合併している28).そして遺伝学的検討はなされていないものの,もやもや病患者の7.9%で腎動脈狭窄が発見されたとの報告もある33).さらにChangらは,末梢性肺動脈狭窄症の5名について検討し,うち4名がRNF213 c.14429G>A p.R4810Kホモ接合性を有し,かつそのうち2名はもやもや病を有していなかったことを確認した34).このように,RNF213 c.14429 G>A p.R4810Kは頭蓋内動脈病変全般のみならず,全身の動脈病変に影響しうる素因バリアントであると考えられる.前述のPAH原因17遺伝子のひとつとしても挙げられているCAV1(Caveolin-1.細胞膜上で陥凹構造を示し,さまざまな受容体が集積しているカベオラ膜の主成分)の血中濃度が,もやもや病患者では低下しているとの報告も,PAHともやもや病の関連を考える上で大変興味深い35).この“RNF213血管障害”研究の,さらなる進展が望まれる.
肺動脈性肺高血圧症の臨床における遺伝学的研究のインパクト
原因遺伝子や修飾遺伝子の候補が次々と同定されるのは喜ばしいことなのだが,問題はこれらの遺伝子のバリアントがPAHの臨床像とどう関わるのか,そしてPAH診療に寄与するのかどうかである.上記のABCC8バリアント,PTGISバリアント,FHITバリアントを保有しているPAH症例に対するジアゾキサイド,イロプロスト,エンザスタウリンの効果のさらなる解明と臨床応用には大いに期待したい.しかし,遺伝学的研究の結果を臨床の場で有効に活用するには,もうしばらく時間を要すると考えられる.前回の総説5)で,PAHにおける遺伝子変異と予後との関係については,筆者らの検討36),英国で実施されたメタアナリシス37)も含めていくつか紹介したが,その後は目ぼしい検討はなされていない.原因遺伝子の数が増えたものの,各々の患者数が少なく,臨床的特徴が曖昧であるためであろう.
小児PAHにおいて,原因遺伝子の内訳はどうなっているのか.2018年に米国から,小児PAH患者では成人発症PAH患者と比較してTBX4変異を持つものが有意に多いとの報告がなされた38).この報告を受けて,筆者らも東京女子医科大学において遺伝学的検査を実施したPAH症例について予備検討を行った.BMPR2バリアントが原因遺伝子として最も多く検出されるというのは他の検討と同様である.しかし,TBX4バリアントが小児で多いということはなく,むしろACVRL1バリアントが小児で多く検出されている(未発表).小児PAHの原因遺伝子の分布は人種や地域で異なるのか,果たしてそれは小児PAHの予後に影響するのか.筆者らは国内のみでの検討には限界があり,国際的な検証が必要と考えている.前述の予備検討結果を踏まえて,現在検証準備を進めている段階にある.
最後に,個人的に最近強く興味を惹かれている,PAH基礎研究の論文を2報紹介する.ひとつめはPAH患者の肺動脈における解糖系の亢進に注目した,2019年のCaoらの報告である39).解糖調節因子である6-phosphofructo-2-kinase/fructose-2,6-bisphosphatas(PFKFB3)を阻害する3POを投与すると,SU5416/低酸素PHモデルラットにおいて肺高血圧の発症がほぼ完全に抑制されることが明らかになった.さらに内皮細胞特異的Pfkfb3ノックアウトマウスを作製し,これを用いて低酸素誘発性PHモデルを作製しようとしたところ,野生型マウスと比較してPHの進行が遅いこと,もしくはPHを発症できないこと,さらには肺動脈平滑筋細胞の増殖能が低下していることが判明した.また,低酸素状態のヒト肺動脈内皮細胞にsiPFKFB3をtransfectionすると,HIF-1αおよびHIF-2αの発現量が有意に低下することも明らかとなった(Fig. 5).もう1報は2018年のDaiらの検討である.この報告ではHIF-2α阻害剤コンパウンド76によるHIF-2αの薬理学的阻害が複数のPAHモデル動物において収縮期右室圧と右室肥大を改善させ,右心不全や右室の線維化,ならびに閉塞性肺血管リモデリングを抑制することが示された40).さらに,モノクロタリン暴露PAHモデルラットに対して,モノクロタリン投与の2週間後にコンパウンド76を投与したところ,右室収縮期血圧,右室肥大,肺血管リモデリングが逆転して右心不全の発症が防止され,生存率が向上したという.
HIF-2αはその下流にBMPR2, IL-6, CXCL12, CAV1等が関連する,既にPAH発症への関与が報告されている複数のシグナル伝達経路を標的としている.これまでPAHの発症メカニズムに関与しうるシグナル伝達経路がいくつも報告されてきたが,HIF-2α阻害剤はこれらの複数のシグナル伝達経路を介してPAHを改善する可能性を秘めている(ちなみに前述のMorikawaらの報告では22),ATOH8は低酸素状態に反応してHIF-2αの誘導を減少させることも明らかとなっている).さまざまな原因遺伝子候補や発症機序についての学説が飛び交い,混沌としたこの研究分野の中で,これらの2報はPAHの病態解明のパズルを完成させるのに不可欠なピースになると,筆者としては考えている.プロスタサイクリン経路,エンドセリン経路,一酸化窒素経路の3系統からのアプローチでは十分な治療が困難である重症PAH症例に対して,このように多系統から一挙に斬りこむことが可能な薬剤の開発が今後は期待される.