小児循環器疾患における基礎研究の発展と展望Development and Prospect of Basic Research in Pediatric Cardiovascular Disease
慶應義塾大学医学部小児科学教室Department of Pediatrics, Keio University School of Medicine ◇ Tokyo, Japan
発行日:2020年6月1日Published: June 1, 2020
先天性心疾患は,その臨床的重要性にもかかわらず,大多数の症例の遺伝的病因は未知であり,いわゆる「多因子性」疾患と考えられてきた.心臓の形態は,複雑な3次元構造を構築するために必要となる,時間的,空間的な遺伝子制御を受けて発生する.先天性心疾患の病因を解明するために,心血管形態形成における個々の部位特異的な発生に焦点を当てたアプローチが重要となる.近年の目覚ましい分子生物学の発展に伴い,遺伝子改変動物,さらには幹細胞を用いた研究が精力的に行われ,心臓の各コンポーネントの発生は複数種の心臓前駆細胞が寄与することが明らかとなり,この20年間で心臓発生および疾患発症メカニズムに関する新たな知見が急速に集積された.このレビューでは,小児循環器疾患の近年の基礎的知見の発展と,今後の展望について記載する.
Despite its clinical importance, the detailed genetic etiology of congenital heart disease is unknown, and it has been considered a “multifactorial” disease. The development of the heart is regulated by the spatiotemporal control of many genes in order to construct a three-dimensional complex form. To elucidate the pathogenesis of congenital heart disease, it is essential to promote research focusing on site-specific development in cardiovascular morphogenesis. Studies using genetically modified animals and stem cells are actively being conducted, and new insights into cardiac development and mechanisms of pathogenesis have accumulated over the past 20 years. These include the detailed roles of multiple cardiac progenitors contributing to the development of each component of the heart. This review describes the developments and future prospects in the field of basic research on pediatric cardiovascular disease.
Key words: cardiac development; iPS cells; cardiac progenitor cells; congenital heart diseases; cardiomyopathy
© 2020 特定非営利活動法人日本小児循環器学会© 2020 Japanese Society of Pediatric Cardiology and Cardiac Surgery
小児循環器領域の基礎医学におけるミッションは,心臓形成機序の理解に基づく,疾患発症メカニズムの解明がその大きなウエイトを占める.基礎医学で得た知見をもとに小児循環器疾患の診断,治療,さらには発症予防を実現することは,小児循環器領域に携わる医療従事者としての悲願である.心臓は胚に形成される最初の臓器であり,生物の生命におけるその後のすべての事象は,生物が生存するうえで必要な酸素と栄養素の需要に十分な血液を循環させる心臓の能力に依存する.心臓の機能は構造の影響を強く受けるが,その形態形成過程には複雑な遺伝子制御機構が必要となる.調節遺伝子に生じた変異は,その形態形成に障害を及ぼし,先天性心疾患(congenital heart diseases: CHD)を引き起こす.CHDの発症メカニズムを解明し,治療法を開発するためには,組織および細胞レベルでの発生異常を明らかにすることが重要となる.心臓発生の正常と異常の理解が,小児循環器領域の疾患発症メカニズム解明のための基礎研究のスタート地点である.
細胞系譜を追跡する技術の開発は,心臓発生起源の特定に大幅な進展をもたらした.心臓原基は,マウス胚の胎生(E)7.5日(人における妊娠2週目まで)に胎仔前方に三日月形(cardiac crescent)を形成する(Fig. 1).この側板中胚葉に由来する心臓前駆細胞の三日月型プールは一次心臓領域(first heart field: FHF)と呼ばれ1, 2),FHFは最終的に左心室と原始心房を形成する.マウスではE8.0,ヒトでは妊娠3週に,この心臓前駆細胞群は腹側正中線上で融合し,心内膜細胞の内層と心筋細胞の外層とからなる原始心筒を形成する.二つの層の間には相互シグナリングを介在する心ゼリーと呼ばれる基質が存在する.
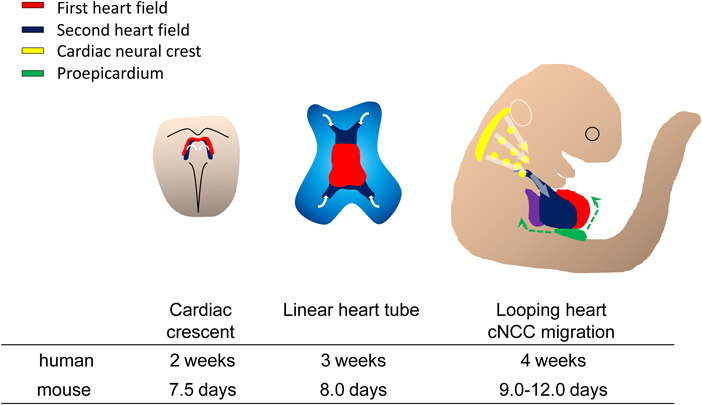
First heart field progenitor cells (red) of the cardiogenic mesoderm forms cardiac crescent under in the anterior part of the embryo at E7.5, then move ventrally to the midline and form the linear heart tube. The second heart field (blue) are situated in medial splanchnic pharyngeal mesoderm and migrate into anterior and posterior part of liner heart tube, providing the source of right ventricle, outflow tract and atrial cardiomyocytes. After the looping of the heart tube around E8.5, cardiac neural crest cells migrate from the dorsal neural tube to III, IV and VI pharyngeal arch arteries and contribute to vascular smooth muscle cells of the aortic arch and cardiac outflow tract around E10.5. At the same time, the proepicardial organ progenitor cells contact the surface of the developing heart, give rise to the epicardium.
その後2001年に,複数の研究グループから,FHFとは異なる,第2の心臓前駆細胞集団の存在が報告された3–6).この心臓前駆細胞集団は二次心臓領域(second heart field: SHF)と呼ばれ,臓側中胚葉に由来する.SHFはcardiac crescentの外側に存在し,次いで原始心筒の後方に位置するようになる.SHF由来心臓前駆細胞はその後原始心筒の動脈側および静脈側より原始心筒内に流入する.原始心筒が右回りにルーピングする時期には,SHF由来心臓前駆細胞は心臓流出路および右心室,両側心房の大部分の心筋を構成する.また,SHF由来の心筋細胞が供給されることにより心臓流出路の伸長が生じる.この過程は,両大血管が左右心室と正しく整列するための心臓流出路のローテーションに必須である.
心臓前駆細胞の第3の系統となる,心臓神経堤細胞(caridac neural crest cells: cNCCs)は,中耳プラコードと第3体節の尾側境界との間の背側神経管に由来する7).背側神経管から剥離した後,cNCCsは尾側咽頭弓から第3, 4および6咽頭弓に遊走し,大動脈弓の形成に参加する.さらに一部のcNCCsは心臓流出路に到達し,流出路中隔を形成する.cNCCs–SHF間の相互作用は,心臓流出路および大動脈弓の発達に不可欠であり,複数のシグナル伝達経路が,cNCCsの遊走および流出路中隔を形成するための集塊形成を制御する8).
心臓前駆細胞の第4の系統は,発生初期胚の心臓と肝臓の間にできる横中隔間葉組織(septum transversum)から発生する心外膜前駆組織(proepicardium: PE)である9).心外膜および冠動脈の細胞の一部は,PEから生じる.PEは胎児心臓に向かって伸長,付着し,心筋表面に広がった後に心外膜を形成する.心外膜形成過程で,一部のPEおよび心外膜細胞が上皮-間葉転換を受け,心外膜下組織から心筋層に入り,冠動脈平滑筋および線維芽細胞へと分化する10)(Fig. 1).
心筋細胞系譜の運命決定を制御する因子の解明に関して,注目すべきブレイクスルーは,ショウジョウバエの原始的な心臓の形成に必要とされるホメオボックス遺伝子tinman(tin)の発見であった11, 12).tinmanが欠失したショウジョウバエ胚は心臓が欠如する.一方,tinmanのhomologであるNkx2.5が欠損したマウスは,心臓の初期発生は正常に起こるが,原始心筒の異常形態形成および左心室発達不全により胎生致死となる13).この報告は,マウスにおけるNKX2.5は,ショウジョウバエ胚におけるtinmanと異なり,他の遺伝子群が相補的な作用を示すことにより,心臓系譜の決定に複雑に関与する可能性を示すものであり,その後多くの研究により,心臓発生過程に必須の心臓転写因子群による複雑な遺伝子制御ネットワークが明らかになりつつある(Fig. 2).
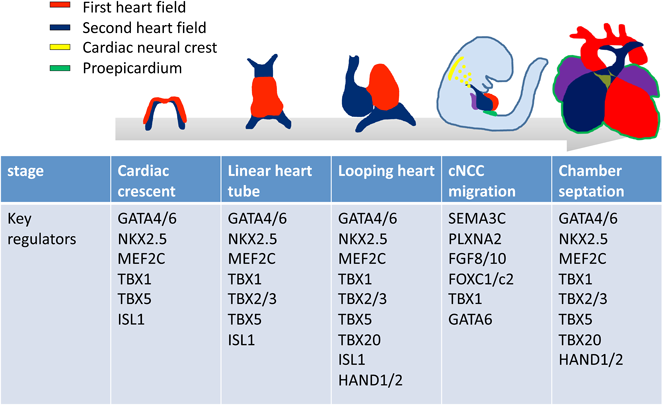
NKX2.5が全心臓前駆細胞に発現するのに対し,LIM-ホメオドメイン転写因子Islet1(ISL1)は初期発生過程でSHFに発現し,SHF系譜の決定における役割を果たす.MADS-box factor myocyte enhancer factor-2(MEF2)は,他の転写因子と協同して,筋原線維タンパク質をコードする遺伝子の発現を直接的に活性化する.脊椎動物に存在する四つのMef2遺伝子(Mef2a, -b, -cおよび-d)の中で,Mef2cは心筋収縮タンパク質の発現および心室構造の発達に必要である14).Mef2c欠損マウスは,SHFの発生障害による右心室低形成を呈する.ジンクフィンガー転写因子のGATAファミリーのメンバーは,脊椎動物において6種存在する.GATAファミリーのうち,GATA4/5/6はNKX2.5, MEF2等の心臓転写因子と協調的に,多数の心筋収縮タンパク質をコードする遺伝子の発現を直接制御し,FHFおよびSHFの発生の中心的役割を果たす15).T-box(Tbx)遺伝子ファミリーは,心臓系譜細胞の誘導,分化および形態形成を制御する16).Tbx5を欠損したマウスは,FHFの発生が障害され,心臓流入路側の重度の低形成を示し胎生致死となる17).TBX20は機能心筋の誘導および増殖に主要な役割を果たし,心室形成に関与する18).TBX2およびTBX3は,TBX20と拮抗的に作用し,刺激伝導系の発生に寄与する19).22q11.2領域に含まれるT-box型転写因子TBX1は,心臓および頭蓋顔面疾患の主要な表現型を決定する疾患原因遺伝子として同定された.TBX1はSHFに発現し,Tbx1欠失マウスは22q11.2欠失症候群(22q11DS)と同様の心血管表現型を有する20).心臓流出路におけるTBX1発現量の調節は,folkhead型転写因子群FOXA2, FOXC1, FOXC2が重要な役割を果たす21, 22).bHLH型転写因子のHANDファミリーのHAND1はFHFに有意な発現を示し,Hand1欠損マウスでは,変異胚は原始心筒の外側湾曲が障害される.HAND2は,主に右心室および心臓流出路に発現し,Hand2欠損マウスは,右心室の重度の形成不全をきたす23).
cNCCsは,さまざまなシグナル因子と相互作用し,これらの分泌因子に近接する多くの構造の形成に関与する.近年注目されているcNCCsの心臓流出路への遊走を制御する因子の一つにSemaphorin 3C(SEMA3C)が挙げられる24).胎生期総動脈幹の肺側心筋はSEMA3Cを発現するが,cNCCはその受容体PlexinA2(PLXNA2)を発現し,SEMA3Cの濃度勾配に従って引き寄せられる.このシグナリングは,心臓流出路へのcNCCsの正確な遊走に必要である25, 26).
心臓流出路の発生にはSHFおよびcNCCsの正常な発生および相互作用が必須となる.そのため,それら二つの心臓前駆細胞の発生制御因子は心臓流出路発生異常の責任遺伝子となる可能性がある.実際にSHFおよびcNCCsの発生制御機構を解析した一連の研究例を以下に示す.
発生制御機構の解析において,疾患原因遺伝子の解析はヒト発生異常疾患の病因に関連した強力なエビデンスを提供しうる.遠隔期予後が問題となる重症心臓流出路異常を有する患者DNAを用いた遺伝子変異解析を行った研究では27, 28),転写因子群の中でも特にGATA転写因子ファミリーのGATA4およびGATA6に多くの変異が同定された.GATA4/6は,心臓発生に必須であり,マウスにおけるGata6のノックアウトは,内胚葉分化の欠陥による初期胚の致死をもたらす.Gata4およびGata6ヌル対立遺伝子のヘテロ接合体マウスは,子宮内で死亡し,心室中隔および流出路中隔欠損を含む総動脈管遺残(PTA)のスペクトルを示す29).これらの実験動物を用いた先行研究の結果は,GATA6が心臓形成に必須の遺伝子群の発現を誘導するため,GATA4と協同して機能する可能性を示している.
GATA6遺伝子変異の影響を確認するために,次に我々は2例のPTA患者に認められた変異の機能解析を行った27)(Fig. 3A).1例の両親は変異,心疾患ともに認めず,発端者はde novo変異であることが判明した.またもう1例では,肺動脈弁の構造異常を含むCHDを有する父および姉妹に発端者と同一の変異が認められ,心疾患を有さない母には変異は存在せず,遺伝子変異と心疾患の相関が確認された(Fig. 3B).近年,Gata6の神経提細胞特異的欠失マウスの心臓表現型は,PTAを呈し胎生致死をきたすことが報告され,GATA6がcNCCsの発生に機能することが示唆された30).そこで我々は“GATA6がcNCCsの流出路への遊走をコントロールする”という仮説を立て,SEMA3Cシグナルに焦点を当て,検討を行った27).まず,マウス胎仔のGata6, Sema3cおよびPlxna2の発現を確認したところ,それらの遺伝子はすべて心臓流出路に発現することが確認された.さらにSEMA3C, PLXNA2の5′側上流に,GATA6が直接結合し,遺伝子発現を調節するエンハンサー領域が存在し,GATA6変異体がそれらエンハンサーに対する転写活性を失っていることが確認された.さらにトランスジェニックマウスを用いた検討では,実際の生体内において心臓流出路のSEMA3CおよびPLXNA2発現がGATA転写因子によって制御されていることが示された(Fig. 3C, D).これらの結果から導き出される結論として,GATA6がSEMA3CおよびPLXNA2の発現を上流で直接制御し,その破綻はcNCCsの発生を障害し,ヒト心臓流出路発生異常の原因となると考えられた.
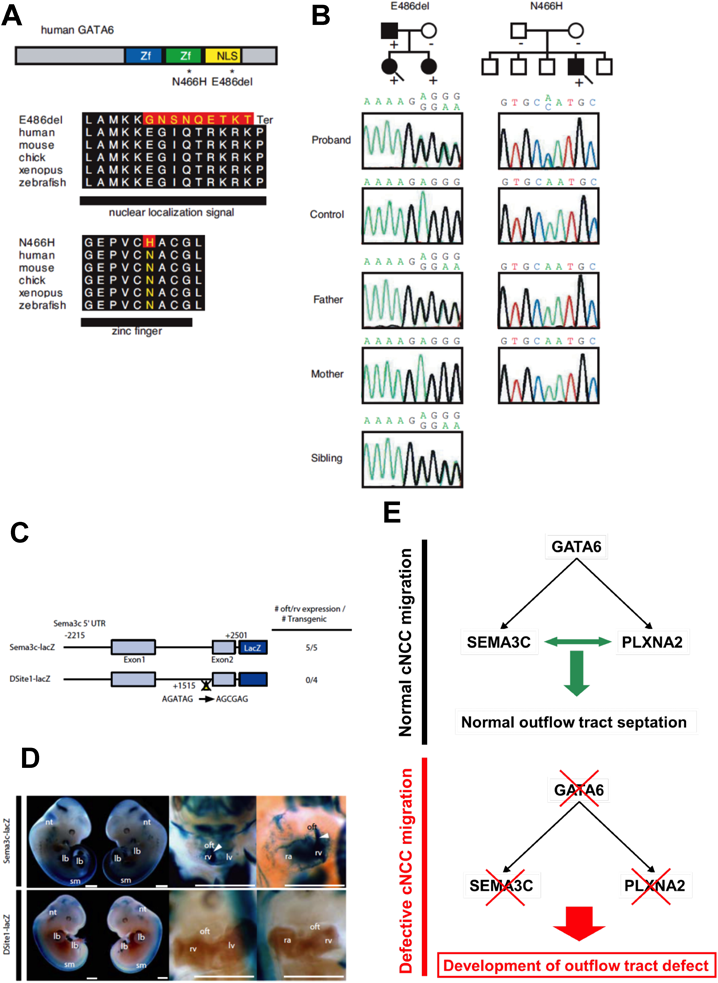
(A) The structure of the human GATA6 gene, the position of the two mutations (asterisks), and conservation of alignment between species are shown. Zf, zinc finger; NLS, nuclear localization signal. Changes in amino acids are highlighted in red. The E486del mutation causes two nucleotide deletions, resulting in nine amino acid changes followed by P489 to termination (Ter) codon. (B) Pedigree indicating cardiac phenotype and the presence (+) or absence (−) of the GATA6 mutation in the family of proband A(E486del; arrow) and proband B(N466H; arrow).The probands are indicated by arrows. (circle), female; (box), male; (solid fill), with CHD. In addition, a sequence chromatogram of one frame shift mutation (E486del, proband A) and a point mutation (N466H, proband B) are shown. (C) GATA cis-elements in the Sema3c enhancer/promoter are essential for their expression in outflow tract development. Schematic diagram of the mouse 4.7-kb Sema3c promoter-lacZ reporter plasmid with or without site-directed mutagenesis of Site1 (Site1-lacZ). The number of transgenic embryos analyzed is indicated. (D) Embryonic day (E) 11.5 embryos harboring the Sema3c-lacZ transgene (left two lanes). Clearing of the embryo (right two lanes) revealed lacZ-positive cells migrating into the outflow tract (oft) and subpulmonary myocardium (arrowheads), and the right ventricle (rv) shown in the higher-magnification image of the heart. No lacZ-positive cells in the oft and rv, shown in the higher-magnification image of the heart in the embryo harboring the transgene with site 1 mutation. sm, somites; lb, limb bud; nt, dorsal neural tube; ra, right atrium; lv, left ventricle; la, left atrium; 3/4, paa 3 and 4; 6, paa 6. (E) The expression of SEMA3C and PLXNA2 in the developing outflow tract is directly regulated by GATA6 through the consensus GATA binding sites. Mutant GATA6 proteins failed to transactivate SEMA3C and PLXNA2, and mutation of the GATA sites on enhancer elements of SEMA3C and PLXNA2 abolished their activity, specifically in the outflow tract/subpulmonary myocardium and cNCC derivatives in the outflow tract region, respectively, suggesting that mutations of GATA6 cause specific forms of human outflow tract defects.
Tbx1欠失マウスの先行研究で,Foxc1/x2欠失マウスと同様に,SEMA3Cの心臓流出路における発現低下が報告された31).そこで22q11.2DSのモデルマウスであるTbx1発現低下マウスを用いて,SEMA3C発現パターンの詳細な変化を検討したところ,咽頭弓領域におけるSEMA3C発現が,Tbx1発現低下マウスでは増強している様子が確認された32)(Fig. 4A).Tbx1発現低下マウスのcNCCsの遊走・分布パターンを観察したところ,野生型に比して心臓流出路領域へ遊走する細胞数が減少する一方で,咽頭弓領域で遊走が停滞し,集塊を作るcNCCsが観察された.TBX1とSEMA3Cの間を介在する因子を検索した結果,TBX1の制御下にSHFより発現,分泌されるFGF8がcNCCsにおけるSEMA3Cの発現を抑制することが明らかとなった.ニワトリ胚とウズラ胚のキメラを用いたex ovoの実験系において,咽頭弓領域にFGF8の中和抗体を注入したキメラ胚では,cNCCsが心臓流出路まで到達せず,FGF8抗体の注入部位でその遊走が停滞する様子が観察された(Fig. 4B).また,ニワトリ胚の第IV–VI咽頭弓動脈間にFGF8中和抗体を注入した後,発生を追跡すると,第IV咽頭弓動脈の発育不全が観察された.
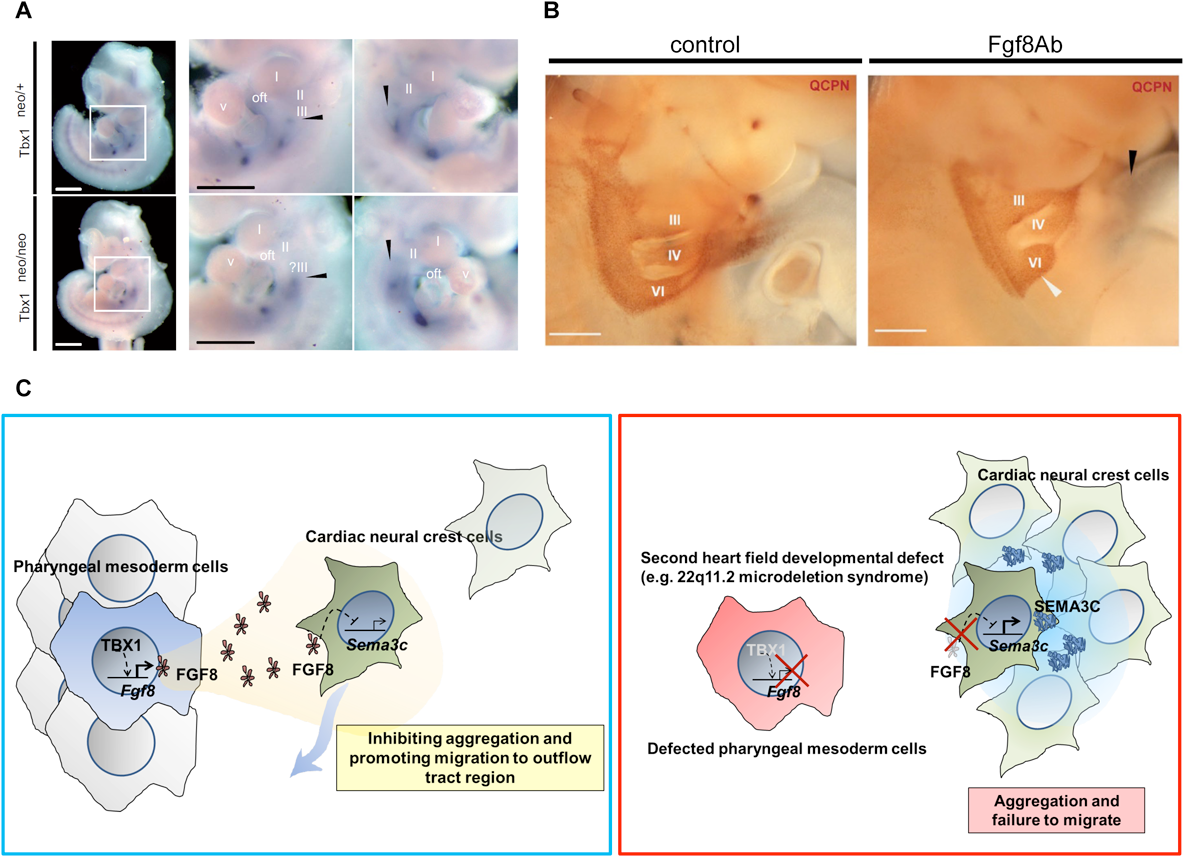
(A) Aberrant mRNA expression of SEMA3C in pharyngeal arch region in the hypomorphic state of TBX1 (Tbx1neo/neo). DIG-labeled whole mount in situ hybridization of SEMA3C mRNA in Tbx1neo/+ (upper, left) and Tbx1neo/neo (lower, left) mouse embryos at E10.5. Higher magnification bilateral views of pharyngeal arch region (white box) are shown in right lane. (B) Pharyngeal arch regions of chick-quail chimeras with bilateral transplantation of cNCCs. Fgf8ab injection causes less migration of cNCCs from the 6th pharyngeal arch region (white arrowhead) to the outflow tract (black arrowhead) compared to control. II, III, IV and VI, 2nd, 3rd, 4th, and 6th pharyngeal arch arteries, respectively. (C) TBX1 in the SHF inhibited the expression of SEMA3C through its downstream signaling molecule, FGF8, in migrating cNCCs along the pharyngeal arches. In contrast, in the embryos with the developmental defect of SHF, downregulation of TBX1-FGF8 signaling in the pharyngeal arch region may lead to misexpression of SEMA3C in migrating cNCCs, resulting in their aggregation and the disruption of migration. SEMA3C is a key signaling molecule that mediates an interaction between cNCCs and the SHF implicated in proper septation of the outflow tract. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ or send a letter to Creative Commons, PO Box 1866, Mountain View, CA 94042, USA.
これらの一連の研究結果よりGATA6は流出路基部におけるSEMA3Cの発現を正に制御する一方で,咽頭弓領域ではTBX1がFGF8を介して,cNCCsの異所性SEMA3C発現を抑制することが明らかとなった.すなわち,SHFにおける心臓転写因子群が,時間的,空間的にSEMA3C発現を制御することにより,cNCCsの適切な咽頭弓動脈および心臓流出路への遊走を制御することが明らかとなった(Fig. 4C).
前述のように,心臓転写因子の重要な機能として,心臓形態発生の制御が挙げられるが,一方で,心筋分化および心室構造の形成にも中心的な役割を果たすことが知られている.しかしこれまで,ヒト心筋細胞の発生を直接観察することは,胎児を研究に用いるという倫理的問題上困難であった.2005年のiPS細胞の開発というブレイクスルーは,これまでのヒト疾患研究の規定概念を変え,患者細胞を使用してヒト幹細胞から体細胞への発生分化過程の異常を再現するという新たな研究戦略を可能とするパラダイムシフトを引き起こした.以下にiPS細胞を利用した,新たな小児循環器領域における研究の方向性を述べる.
発症原因が心室筋形成の発生過程の障害と密接に関連すると考えられる心疾患として,左室心筋緻密化障害(left ventricular non compaction: LVNC)が挙げられる33).心筋緻密化は,肉柱形成,緻密層増殖および肉柱層リモデリングの各プロセスが,時間的・空間的に制御されることにより完成される.これまでにヒトおよび動物を用いた研究で,心筋細胞の細胞周期制御に関わる遺伝子群の変異が胎児心筋細胞の増殖能異常を引き起こし,LVNC様の表現型を呈する例が報告されてきたが,その詳細なメカニズムは未だ不明である34–41).iPS細胞由来心筋細胞(iPS cell-derived cardiomyocyte: iPS-CM)は,患者の遺伝的背景をそのまま保存しており,疾患モデルへの利用が期待される.そこで今回我々は,その病因を明らかにするため,LVNC家族歴を有する家系から疾患特異的iPS細胞を作製した42)(Fig. 5A).LVNC患者由来iPSC-CMsをコントロール群と比較したところ,LVNC群に有意な増殖能低下が確認された(Fig. 5B).LVNC iPSC-CMsに認められた増殖能低下の原因を解明するため,RNAシークエンシングを用いた網羅的遺伝子発現解析を行った結果,Upstream解析でLVNC iPSC-CMsは,コントロール群と比べてTGFβシグナルの活性化が顕著であることが示された.TGFβシグナルの活性化が心筋細胞増殖能に与える影響を調べるため,培地にTGFβ1を添加したところ,iPSC-CMsのS期心筋細胞率は有意に低下し,TGFβが心筋細胞増殖に抑制的に作用する可能性が考えられた.
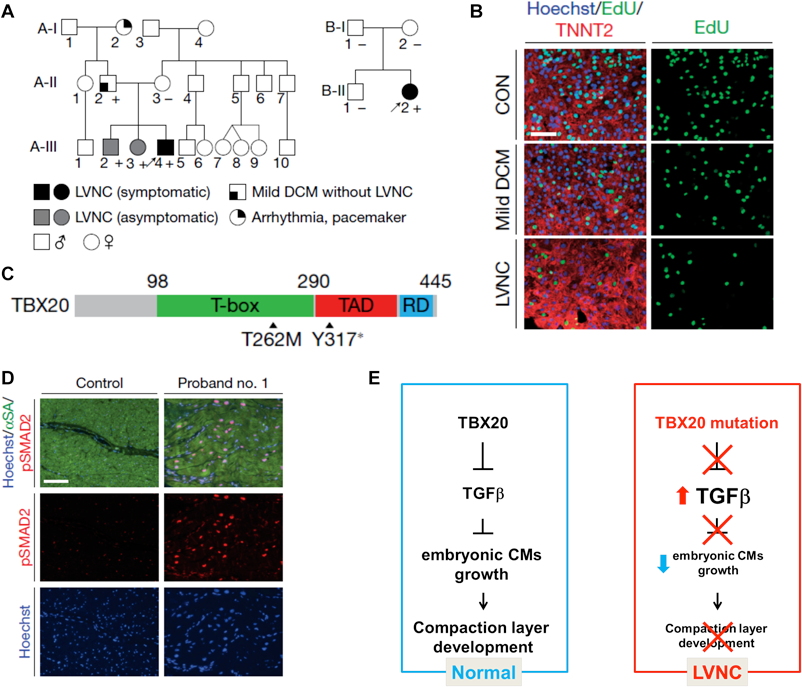
(A) Schematic pedigree of two families with LVNC. The probands are indicated by arrows (A-III-4 and B-II-2). Plus and minus signs indicate the presence and absence of the TBX20 Y317 mutation in the family A and the T262M mutation in the family B, respectively. (B) Immunostaining of nuclear (blue), TNNT2 (red) and EdU (green) in iPSC-CMs derived from the proband, father and healthy control. The iPSC-CMs derived from the proband showed significant decrease of EdU positive cells suggesting proliferation defect in the LVNC iPSC-CMs. (C) Schema of TBX20 and the position of the Y317 and T262M mutations. TAD, transactivation domain; RD repression domain. (D) Immunostaining of nuclear (blue), alpha-sarcomeric actin (green) and phospho-SMAD2 (red) in the LV of control donor heart tissue versus the explanted heart of proband no. 1. (E) Mechanistic schema of the pathology of LVNC caused by TBX20 loss of function mutation. During the myocardium development, TBX20 inhibits activation of TGFβ signaling and promotes embryonic cardiomyocyte proliferation. TBX20 mutation disturbs the suppression of TGFβ and promotes early exit of cardiomyocyte cell cycle. Decreased ventricular cardiomyocyte proliferation leads to failed formation of compact layer of developing myocardium and results in LVNC.
この家系における疾患原因遺伝子の解明のため,エクソームシークエンスを用いて,発端者,同胞および両親の網羅的遺伝子変異解析を行った結果,心臓表現型の有無に一致してTBX20のナンセンス変異が認められた(Fig. 5A, C).TBX20がLVNCの疾患原因遺伝子であることを証明するために,富山大学,市田蕗子先生をはじめとする諸先生方のご協力のもとに,LVNC患者78名のDNAを解析したところ,孤発例1人よりDNA結合領域にde novo点変異が検出され,TBX20変異の病原性が証明された(Fig. 5A, C).TBX20の障害が心筋細胞内でTGFβシグナル活性に与える影響を調べるため,ヒトES細胞のTBX20をノックダウンした細胞株を作製し,心筋分化誘導後に細胞増殖能およびTGFβシグナル関連因子の発現を調べた結果,LVNC iPS-CMと同様にTBX20ノックダウンES細胞由来心筋細胞は,その細胞増殖能は低下し,TGFβ関連遺伝子の発現は著明に増加することが確認され,TBX20遺伝子の機能障害がTGFβシグナル活性化を引き起こす可能性が示唆された.さらにLVNC患者の心臓でTGFβシグナルの活性化が認められることを確認するため,発端者の移植時に摘出した心臓をサンプルとして解析を行ったところ,iPSC-CMsの所見と同様に,発端者心筋ではTGFβシグナル下流のSMADのリン酸化亢進が認められた(Fig. 5D).
次に我々は,TGFβシグナルの活性化が生体内の心筋発生に与える影響を調べるため,心筋細胞特異的にTGFβ1を発現し,発現量をdoxycyclineにより調節可能なトランスジェニックマウスを作製した.TGFβ1を中等度に発現亢進させたマウスは胎生13日までに死亡した.胎生12.5日の心筋を解析したところ,緻密層心筋細胞の増殖が低下し,緻密層が菲薄化していることが明らかとなった.
最後に治療へ関連する知見として,LVNC iPSC-CMsの表現型を救済するため,TGFβシグナルの修正,およびDNAレベルでの修復を試みた.TGFβ受容体の特異的な阻害作用を有する低分子化合物を用いてTGFβシグナルを阻害することにより,LVNC iPSC-CMsの細胞増殖能は改善した.さらに,発端者由来iPS細胞にゲノム編集を行い,TBX20変異配列を正常配列で置換したところ,LVNC iPSC-CMsの増殖能低下は改善が認められ,TBX20変異がLVNC iPSC-CMsの表現型に主たる影響を与えていることが確認された.以上の結果をまとめると,TBX20はTGFβシグナルを抑制し,胎児心筋増殖を促し,緻密層形成に貢献するが,TBX20遺伝子変異によりTBX20の機能が損なわれると,TGFβシグナルの異常活性化が惹起される.TGFβシグナルの亢進は胎児心筋増殖を障害し,緻密層形成不全が引き起こされ,LVNCが発症すると考えられた(Fig. 5E).従来の遺伝子変異解析と動物モデルに加えて,ヒト組織および患者由来iPS細胞より心臓構成細胞を作製し,表現型解析を行うことにより,ヒトにおける心臓発生過程の異常に際しても,詳細な病因解明および薬剤開発への足掛かりとなる有用な所見が得られる可能性がある.今後このような手法により,多くのCHDの病態解明,治療法開発が発展することが期待される.
この20年間,分子生物学的手法や遺伝情報処理,幹細胞技術の発展によりその秘密を明らかにするための知見が急速に集積され,多様な心臓前駆細胞の系譜が特定されてきたが,実際に心臓がその複雑で統合された構造を形成するのに対し,前駆細胞集団がどのように寄与するかは,解明すべき点が多く残される領域である.心臓生物学および分子医学の将来の進歩は,複数の分野のインターフェースに新しい技術を統合することによって推進される.心臓病関連遺伝子のマッピングは,ヒトゲノム配列の完成によって促進されてきた.心疾患の表現型に影響を与える修飾因子および遺伝的多型を同定する能力は,疾患の発症機序についての洞察を提供し,テイラーメイド療法の開発に向けた具体的な道しるべとなった.疾患モデル生物における遺伝子を効率的に不活性化する技術は,心臓機能および機能不全を制御する遺伝子の機能を迅速に解明する戦略をもたらした.洗練された新しいイメージング法は,細胞下,細胞内,および器官全体のレベルでの心機能への遺伝的および化学的影響の分析を可能にしつつある.このような複合的な多分野の技術統合よってもたらされる強力で新しいアプローチをもとに,遺伝子治療や幹細胞再生医療の技術をさらに最適化することにより,心臓発生領域の基礎研究は,治療,再生,そして予防への道を切り開く魅力的な可能性を我々の前に示し続けている.
日本小児循環器学会の定める,利益相反に関する開示項目はありません.
1) Buckingham M, Meilhac S, Zaffran S: Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet 2005; 6: 826–835
2) Srivastava D: Making or breaking the heart: From lineage determination to morphogenesis. Cell 2006; 126: 1037–1048
3) Kelly RG, Brown NA, Buckingham ME: The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev Cell 2001; 1: 435–440
4) Waldo KL, Kumiski DH, Wallis KT, et al: Conotruncal myocardium arises from a secondary heart field. Development 2001; 128: 3179–3188
5) Cai CL, Liang X, Shi Y, et al: Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell 2003; 5: 877–889
6) Mjaatvedt CH, Nakaoka T, Moreno-Rodriguez R, et al: The outflow tract of the heart is recruited from a novel heart-forming field. Dev Biol 2001; 238: 97–109
7) Hutson MR, Kirby ML: Model systems for the study of heart development and disease. Cardiac neural crest and conotruncal malformations. Semin Cell Dev Biol 2007; 18: 101–110
8) Waldo KL, Hutson MR, Stadt HA, et al: Cardiac neural crest is necessary for normal addition of the myocardium to the arterial pole from the secondary heart field. Dev Biol 2005; 281: 66–77
9) Mikawa T, Gourdie RG: Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev Biol 1996; 174: 221–232
10) Olivey HE, Svensson EC: Epicardial-myocardial signaling directing coronary vasculogenesis. Circ Res 2010; 106: 818–832
11) Bodmer R: The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development 1993; 118: 719–729
12) Azpiazu N, Frasch M: Tinman and bagpipe: Two homeo box genes that determine cell fates in the dorsal mesoderm of Drosophila. Genes Dev 1993; 7 7B: 1325–1340
13) Lyons I, Parsons LM, Hartley L, et al: Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev 1995; 9: 1654–1666
14) Olson EN: Gene regulatory networks in the evolution and development of the heart. Science 2006; 313: 1922–1927
15) Sorrentino RP, Gajewski KM, Schulz RA: GATA factors in Drosophila heart and blood cell development. Semin Cell Dev Biol 2005; 16: 107–116
16) Plageman TF Jr., Yutzey KE: T-box genes and heart development: Putting the “T” in heart. Dev Dyn 2005; 232: 11–20
17) Bruneau BG, Nemer G, Schmitt JP, et al: A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 2001; 106: 709–721
18) Brown DD, Martz SN, Binder O, et al: Tbx5 and Tbx20 act synergistically to control vertebrate heart morphogenesis. Development 2005; 132: 553–563
19) Moorman AFM, Christoffels VM: Cardiac chamber formation: Development, genes, and evolution. Physiol Rev 2003; 83: 1223–1267
20) Garg V, Yamagishi C, Hu T, et al: Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Dev Biol 2001; 235: 62–73
21) Yamagishi H, Maeda J, Hu T, et al: Tbx1 is regulated by tissue-specific forkhead proteins through a common Sonic hedgehog-responsive enhancer. Genes Dev 2003; 17: 269–281
22) Maeda J, Yamagishi H, McAnally J, et al: Tbx1 is regulated by forkhead proteins in the secondary heart field. Dev Dyn 2006; 235: 701–710
23) Yamagishi H, Yamagishi C, Nakagawa O, et al: The combinatorial activities of Nkx2.5 and dHAND are essential for cardiac ventricle formation. Dev Biol 2001; 239: 190–203
24) Toyofuku T, Yoshida J, Sugimoto T, et al: Repulsive and attractive semaphorins cooperate to direct the navigation of cardiac neural crest cells. Dev Biol 2008; 321: 251–262
25) Brown CB, Feiner L, Lu MM, et al: PlexinA2 and semaphorin signaling during cardiac neural crest development. Development 2001; 128: 3071–3080
26) Feiner L, Webber AL, Brown CB, et al: Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development 2001; 128: 3061–3070
27) Kodo K, Nishizawa T, Furutani M, et al: GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc Natl Acad Sci USA 2009; 106: 13933–13938
28) Kodo K, Nishizawa T, Furutani M, et al: Genetic analysis of essential cardiac transcription factors in 256 patients with non-syndromic congenital heart defects. Circ J 2012; 76: 1703–1711
29) Xin M, Davis CA, Molkentin JD, et al: A threshold of GATA4 and GATA6 expression is required for cardiovascular development. Proc Natl Acad Sci USA 2006; 103: 11189–11194
30) Lepore JJ, Mericko PA, Cheng L, et al: GATA-6 regulates semaphorin 3C and is required in cardiac neural crest for cardiovascular morphogenesis. J Clin Invest 2006; 116: 929–939
31) Theveniau-Ruissy M, Dandonneau M, Mesbah K, et al: The del22q11.2 candidate gene Tbx1 controls regional outflow tract identity and coronary artery patterning. Circ Res 2008; 103: 142–148
32) Kodo K, Shibata S, Miyagawa-Tomita S, et al: Regulation of Sema3c and the interaction between cardiac neural crest and second heart field during outflow tract development. Sci Rep 2017; 7: 6771
33) Kohli SK, Pantazis AA, Shah JS, et al: Diagnosis of left-ventricular non-compaction in patients with left-ventricular systolic dysfunction: Time for a reappraisal of diagnostic criteria? Eur Heart J 2008; 29: 89–95
34) Kosaka Y, Cieslik KA, Li L, et al: 14-3-3epsilon plays a role in cardiac ventricular compaction by regulating the cardiomyocyte cell cycle. Mol Cell Biol 2012; 32: 5089–5102
35) Chen Q, Chen H, Zheng D, et al: Smad7 is required for the development and function of the heart. J Biol Chem 2009; 284: 292–300
36) DiMichele LA, Hakim ZS, Sayers RL, et al: Transient expression of FRNK reveals stage-specific requirement for focal adhesion kinase activity in cardiac growth. Circ Res 2009; 104: 1201–1208
37) Bartram U, Molin DG, Wisse LJ, et al: Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation 2001; 103: 2745–2752
38) Shou W, Aghdasi B, Armstrong DL, et al: Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature 1998; 391: 489–492
39) Grego-Bessa J, Luna-Zurita L, del Monte G, et al: Notch signaling is essential for ventricular chamber development. Dev Cell 2007; 12: 415–429
40) Luxan G, Casanova JC, Martinez-Poveda B, et al: Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med 2013; 19: 193–201
41) Arndt AK, Schafer S, Drenckhahn JD, et al: Fine mapping of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am J Hum Genet 2013; 93: 67–77
42) Kodo K, Ong SG, Jahanbani F, et al: iPSC-derived cardiomyocytes reveal abnormal TGF-beta signalling in left ventricular non-compaction cardiomyopathy. Nat Cell Biol 2016; 18: 1031–1042


This page was created on 2020-05-21T14:30:28.483+09:00
This page was last modified on 2020-06-18T10:00:27.000+09:00
このサイトは(株)国際文献社によって運用されています。