症例
2か月 女児
主訴
哺乳不良
家族歴
父が大動脈基部拡大および水晶体偏位を認め,MFSと診断されている.
現病歴
在胎40週0日,体重3,242 g,正常経膣分娩で出生した.日齢3に心雑音を聴取し,心エコーでVSDと診断された.AR, MRはなかった.生後1か月より高肺血流による容量負荷のため利尿剤の内服を開始したが,徐々に哺乳不良,体重増加不良を認めた.生後2か月の心エコーで左室拡大に加え収縮力低下も認めたため,当科を紹介され入院した.
入院時現症
身長59.5 cm(1.3SD),体重4,376 g(−1.4SD),血圧82/50 mmHg,脈拍140回/分,呼吸数60回/分,経皮酸素飽和度97%.
陥没呼吸を認めた.胸骨左縁第四肋間にLevine 3/6の収縮期雑音およびIII音を聴取した.右季肋部に肝を2 cm触知した.軽度の末梢冷感を認めた.眼球陥入,漏斗胸,細長い四肢,指趾を認めた.耳介の変形や関節拘縮はなかった.
検査所見
血液検査
BNP 1569.8 pg/mL
胸部レントゲン
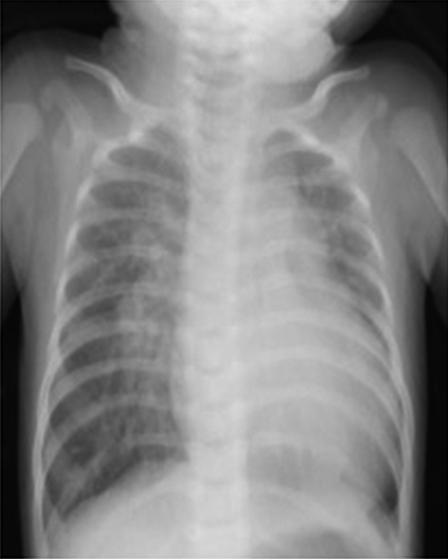
心胸郭比63%,肺血管陰影の増強を認めた(Fig. 1).
心電図
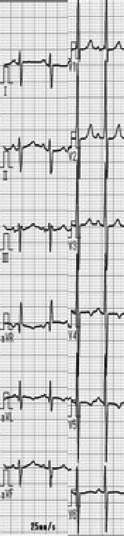
左房負荷,両室肥大を認めた(Fig. 2).
心エコー
左心系の拡大(左室拡張末期径29.8 mm(145% of normal))と収縮能の低下(左室駆出率(LVEF)46%)を認めた.VSDは膜様部欠損で8.1×10.2 mmと大きく,カラードップラーで左右短絡を認め,流速は0.9 m/sと高度の肺高血圧が示唆された.径2.8 mmの心房中隔欠損(ASD)あり,カラードップラーで左右短絡を認めた.軽度の三尖弁逆流を認めた.AR, MRはなかった.大動脈弁輪の拡大はなかった.
経過
入院6日目に心臓カテーテル検査を施行した(Table 1).肺動脈圧67/31(44)mmHg,肺体血流比(Qp/Qs)3.3,肺血管抵抗3.8 u·m2,左室拡張容積(LVEDV)34.4 mL(286% of normal),LVEF 34%,右室拡張容積(RVEDV)29.3 mL(258% of normal),右室駆出率(RVEF)48%と高肺血流による肺高血圧,左室容量負荷,左室収縮能低下を認めた.冠動脈起始異常はなかった.
Table 1 1st and 2nd cardiac catheter data | 1st | 2nd |
|---|
| RA (mmHg) | 3 | 4 |
| mPA (mmHg) | 67/31 (44) | 23/15 (18) |
| LA (mmHg) | 6 | 5 |
| aAo (mmHg) | 88/47 (61) | 86/42 (57) |
| CI (L/min/m2) | 3.1 | 3.2 |
| Qp/Qs | 3.3 | 1.8 |
| Rp (units·m2) | 3.8 | 2.3 |
| LVEDV (% of normal) | 286 | 254 |
| LVEF (%) | 34 | 53 |
| RVEDV (% of normal) | 258 | 228 |
| RVEF (%) | 48 | 57 |
| RA: right atrium, mPA: main pulmonary artery, LA: left atrium, aAo: ascending aorta, CI: cardiac index, Qp/Qs: the ratio of pulmonary to systemic blood flow, Rp: pulmonary vascular resistance, LVEDV: end-diastolic left ventricular volume, LVEF: left ventricular ejection fraction, RVEDV: end-diastolic right ventricular volume, RVEF: right ventricular ejection fraction |
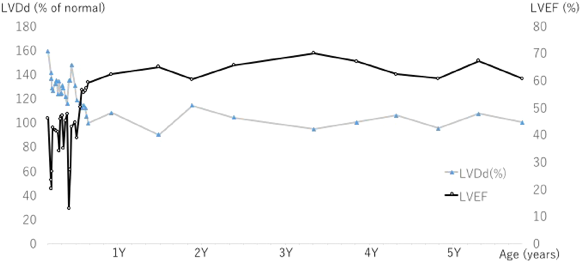
心室中隔欠損が血行動態悪化の主因と考えられ,早期の心内修復術を予定した.Fig. 3に周術期の経過を示す.カテーテル検査後より哺乳量低下,不機嫌などの心不全症状が悪化し,ドブタミン投与を開始した.入院14日目に哺乳不良,多呼吸がさらに増悪し,心エコーで心収縮能が低下し(LVEF 23%),心不全の悪化と判断した.心内修復術は困難と判断し,入院15日目に肺動脈絞扼術(周径:体重+20 mm)を施行した(手術時間:2時間39分).術後は高度の左心不全,低心拍出量症候群を呈した.術後4日に人工呼吸器を離脱,術後6日に集中治療室を退室した.LVEF 40%前後と心収縮能の低下が続いたため,慢性心不全の治療として術後17日よりエナラプリルの内服を0.025 mg/kg/dayより開始し,0.1 mg/kg/dayまで漸増した.LVEFは著変なかったが,心不全症状は徐々に改善した.
生後3か月半,術後41日に心臓カテーテル検査(Table 1)を施行した.肺動脈圧23/15(18)mmHg, LVEDV 32.1 mL(254% of normal),LVEF 53%, RVEDV 27.2 mL(228% of normal),RVEF 57%, Qp/Qs 1.8であり,心内修復術の方針とした.生後5か月時にVSDおよびASD閉鎖を施行した(手術時間:6時間23分,体外循環時間:2時間20分,心停止時間:56分).初回手術と同様に術後は高度の左心不全,低心拍出量症候群を認めた(Fig. 3).術後4日に人工呼吸器離脱,術後12日に集中治療室を退室した.また,手術と同時に施行した心筋生検では,心筋細胞の肥大,心筋細胞間の軽度の線維の増生および配列の乱れを認めたが特異的な所見ではなかった(Fig. 4).心不全が続くため,術後27日よりカルベジロール(0.1 mg/kg/day)とジゴキシン(0.01 mg/kg/day)の投与を開始した.カルベジロールは0.2 mg/kg/dayまで漸増した.LVEFは徐々に改善した.生後7か月(術後85日)に退院した.
以降は外来で経過観察されており,経過をFig. 5に示す.心機能は術後2か月にはほぼ正常化し,以降は6歳になった現在まで増悪はない.現在はカルベジロール(0.2 mg/kg/day),ロサルタン(1 mg/kg/day)の内服を継続している.
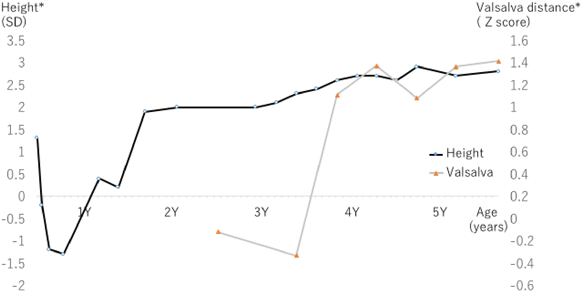
身長の推移,大動脈Valsalva径の推移をFig. 6に示す.徐々に高身長が目立つようになり,また3歳半ころより徐々にValsalvaの拡大を認めるようになった.心外合併症としては,入院時に眼球陥入,漏斗胸,細長い四肢・指趾が認められたのに加え,3歳時に水晶体脱臼,側弯を指摘された.水晶体脱臼およびMFSの家族歴から改訂Ghentの診断基準を満たした.家族の同意を得られず,遺伝子検査は施行していない.
Marfan症候群(MFS)は,全身の結合組織に存在するfibrillin-1をコードする遺伝子FBN1の異常を原因とした常染色体優性の遺伝子疾患である.骨格,目,心臓,大動脈,肺などの多くの臓器に対して様々な程度の障害を起こす症候群である.本症例では遺伝子検査は施行していないが,MFSの家族歴と水晶体脱臼から改訂Ghent基準1)を満たしたのでMFSと診断した.
本症例の鑑別診断には先天性拘縮性クモ指症(CCA)や新生児MFS(nMFS)が上がる.CCAは,高身長やクモ状指趾などMFSと類似した身体的特徴を持つ.幼少期発症のlethal CCAではVSD, ASD,大動脈離断症(IAA)などの先天性心疾患を合併することがある2).しかし,CCAではMFSと異なり出生時より折れ曲がった耳と関節拘縮を認めることが多い一方,水晶体偏位,高度近視,進行性の大動脈基部の拡張を合併することは少ない3).CCAはFBN2遺伝子異常に起因し,遺伝子検査で鑑別する場合もあるが,本症例では臨床所見より否定的と判断した.nMFSは,一般的なMFS(classical Marfan syndrome: cMFS)と同様にFBN1遺伝子の異常によるfibrillin-1の障害によって,弾性線維の形成に障害をきたすと考えられている4, 5).cMFSの遺伝子変異がFBN1遺伝子のほぼ全体に分布しているのとは対照的に,nMFSの遺伝子変異の報告ではFBN1遺伝子のexon24–32の間の変異が多く,これがcMFSとnMFSとの臨床像の違いとなっていると推測されている5, 6).cMFSで同領域に変異を持つこともあり,その場合は重篤な心血管系の合併症の頻度が高いとされている4, 6, 7).変異の種類としてミスセンス変異,ナンセンス変異,フレームシフト変異,スプライス異常などが報告されており,ミスセンス変異が最も多いが6),変異の種類と臨床症状との関連の報告はない.nMFSに明確な診断基準はないが8–10),一般的にnMFSは重篤な房室弁の逆流,肺気腫が特徴的であり,新生児から乳児期より重篤な心肺機能不全を呈し,極めて予後不良であることが多く11),MFSの身体的特徴を持った新生児の症例とは区別するべきとされている3, 12).本症例ではVSDを合併して重度の心不全となったが,出生時に有意な弁逆流を認めず,また肺病変も認めていない.現在は水晶体脱臼や大動脈弁輪拡大などのMFSに伴う症状を認めるが,心機能は正常化しておりnMFSではないと判断した.
MFSでは僧帽弁の粘液腫様変性や弁輪の拡大によるMRやARが生じやすく,弁逆流の増悪による心不全が広く知られているが,MFSの心筋そのものについても潜在的な障害を示唆した様々な報告がある.収縮障害を示唆する報告としては,3Dエコー13),MRI14–16)で評価したものがある.その中でMFS群は対照群と比しLVEFの有意な低下を認めたが,53~57%13–16)といずれも軽度であった.また心エコーの組織ドップラーでの拡張障害を示唆する報告13, 14, 17, 18)もある.右心機能についてMRIで検討した報告では,左心機能と同様に右心機能の低下を認めたがいずれも軽度15, 16)であり,頻度も低かった16).さらにN末端プロBNP(NT-pro BNP)を217人のMFS患者と正常群で比較した検討17)では,軽度の上昇(70.6±74.8 pg/mL vs 58.4±101.1 pg/mL)を認めた.心移植を受けた心不全を有するMFSの患者を検討し,弁逆流とは別にMFS自体による心筋障害が心不全へ関与したと推察する報告19)もあった.外科的治療後のMFS 421例の心機能を検討した報告20)では11%に心機能障害があり,3%は術前から,8%は術後より低下が認められた.MFSでは術中の心筋虚血に弱い可能性も指摘されている.
fibrillin-1はトランスフォーミング増殖因子(TGF-β)の発現に影響を与えるほか,細胞外マトリックスの不可欠な構成成分として存在する.MFSの症状は主にTGF-βのシグナル異常によるとされているが,細胞外マトリックスの構造異常も心機能障害の要因と考えられている17, 21, 22).心筋の病理所見の報告は少ないが,重度の心不全を呈し心移植を受けたMFS患者の報告23)では,心筋病理では特異的な所見はなく,本症例と同様の所見であった.
本症例ではVSDによる高肺血流,左心容量負荷の所見があったが,有意な弁逆流はなかった.心筋生検で特異的な所見は得られず,血行動態が正常化した現在では明らかな心機能低下はない.しかし,術前に血行動態のみでは説明が困難な高度の収縮障害を認めたこと,二度の手術後に高度の収縮障害を認めたことからは,MFSでは心機能障害が潜在する場合があり,VSDの合併など心負荷がかかった際や周術期において,心機能障害の増悪因子となる可能性が考えられる.
MFSに先天性心疾患を合併することは稀であるが,VSD24–28),大動脈縮窄29),肺動脈弁欠損30),右胸心31)などの合併の報告がある.生後4か月で心不全症状が顕在化したVSDに対し心内修復術が施行されたMFS症例25)では,高肺血流性心不全および高度の肺高血圧であったが,術前,術後ともに心機能低下はなく,本症例の経過とは異なった.この症例の詳細な血行動態評価は不明だが,本症例との違いは,VSDによる心負荷の程度やMFSに起因する心筋障害の程度の違いによるものかもしれない.
引用文献References
1) Loeys BL, Dietz HC, Braverman AC, et al: The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010; 47: 476–485
2) Wang M, Clericuxio CL, Godfrey M: Familial occurrence of typical and severe lethal congenital contractural arachnodactyly caused by missplicing of exon 34 of fibrillin-2. Am J Hum Genet 1996; 59: 1027–1034
3) Booms P, Cisler J, Mathews KR, et al: Novel exon skipping mutation in the fibrillin-1 gene: Two ‘hot spots’ for the neonatal Marfan syndrome. Clin Genet 1999; 55: 110–117
4) Tiecke F, Katzke S, Booms P, et al: Classic, atypically severe and neonatal Marfan syndrome: Twelve mutations and genotype-phenotype correlations in FBN1 exons 24–40. Eur J Hum Genet 2001; 9: 13–21
5) Gavilan C, Herraiz I, Granados MA, et al: Prenatal diagnosis of neonatal Marfan syndrome. Prenat Diagn 2011; 31: 610–613
6) 森崎裕子,森崎隆幸:Marfan症候群—遺伝子変異・治療法についての最近の知見.日小児循環器会誌2009: 25; 594–599
7) Faivre L, Collod-Beroud G, Loeys BL, et al: Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet 2007; 81: 454–466
8) Morse RP, Rockenmacher S, Pyeritz RE, et al: Diagnosis and management of infantile marfan syndrome. Pediatrics 1990; 86: 888–895
9) Edwards RH: Congenital Marfan syndrome. Birth Defects Orig Artic Ser 1975; 11: 329–331
10) Gross DM, Robinson LK, Smith LT, et al: Severe perinatal Marfan syndrome. Pediatrics 1989; 84: 83–89
11) 岩朝 徹:Neonatal Marfan syndromeとinfantile severe Marfan Syndrome. 日小児循環器会誌2014; 30: 78–79
12) Hennekam RC: Severe infantile Marfan syndrome versus neonatal Marfan syndrome. Am J Med Genet A 2005; 139: 1
13) Abd El Rahman M, Haase D, Rentzsch A, et al: Left ventricular systolic dysfunction in asymptomatic Marfan syndrome patients is related to the severity of gene mutation: Insights from the novel three dimensional speckle tracking echocardiography. PLoS One 2015; 10: e0124112
14) De Backer JF, Devos D, Segers P, et al: Primary impairment of left ventricular function in Marfan syndrome. Int J Cardiol 2006; 112: 353–358
15) de Witte P, Aalberts JJ, Radonic T, et al: Intrinsic biventricular dysfunction in Marfan syndrome. Heart 2011; 24: 2063–2068
16) Alpendurada F, Wong J, Kiotsekoglou A, et al: Evidence for Marfan cardiomyopathy. Eur J Heart Fail 2010; 12: 1085–1091
17) Gehle P, Robinson PN, Heinzel F, et al: NT-proBNP and diastolic left ventricular function in patients with Marfan syndrome. Int J Cardiol Heart Vasc 2016; 12: 15–20
18) Savolainen A, Nisula L, Keto P, et al: Left ventricular function in children with the Marfan syndrome. Eur Heart J 1994; 15: 625–630
19) Knosalla C, Weng YG, Hammerschmidt R, et al: Othotopic heart transplantation in patients with Marfan syndrome. Ann Thorac Surg 2007; 83: 1691–1695
20) Hetzer R, Siegel G, Delmo Walter EM: Cardiomyopathy in Marfan syndrome. Eur J Cardiothorac Surg 2016; 49: 561–567, discussion, 567–568
21) Cook JR, Carta L, Benard L, et al: Consortium: Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J Clin Invest 2014; 124: 1329–1339
22) Neptune ER, Frischmeyer PA, Arking DE, et al: Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet 2003; 33: 407–411
23) 伊藤愛剛,柴 信行,小丸達也:心臓移植を施行したMarfan症候群の1例.心臓2006; 38: 958–963
24) Roberts WC, Honig HS: The spectrum of cardiovascular disease in the Marfan syndrome: A clinico-morphologic study of 18 necropsy patients and comparison to 151 previously reported necropsy patients. Am Heart J 1982; 104: 115–135
25) Rozendaal L, Blom NA, Hilhorst-Hofstee Y, et al: Dilatation of the great arteries in an infant with Marfan syndrome and ventricular septal defect. Case Rep Med 2011; 4: 172109
26) Nathan D, Kraus J, Deutsch V, et al: Dilatation of the aorta and pulmonary artery with aortic and pulmonary insufficiency in the presence of a ventricular septal defect and infundibular pulmonic stenosis: Report of a case of forme fruste of the Marfan syndrome. Dis Chest 1966; 50: 199–205
27) Yajnik VH, Kshatriya PK, Vaidya MH, et al: Marfan’s syndrome, ventricular septal defect and basilar impression in one patient. J Indian Med Assoc 1975; 64: 242–244
28) Bilgrami NL, Tewari SG, Aslam M, et al: Marfan syndrome with microcornea, aphakia and ventricular septal defect: Case report. Indian Heart J 1981; 33: 78–80
29) Headley RN, Carpenter HM, Sawyer CG: Unusual features of Marfan’s syndrome including two postmortem studies. Am J Cardiol 1963; 11: 259–266
30) Childers RW, Mccrea PC: Absence of the pulmonary valve: A case occurring in the Marfan syndrome. Circulation 1964; 29: 598–603
31) Metelka R, Dusek J, Vomacka J: Dextrocardia and Marfan’s syndrome. Acta Univ Palacki Olomuc Fac Med 1992; 133: 43–45