肺高血圧(PH)は,安静時の平均肺動脈圧が≧25 mmHgという定義が,国際的に,小児成人を問わず使用されてきた1).最近のThe 6th World Symposium on Pulmonary Hypertensionでは,成人PHにおいて平均肺動脈圧>20 mmHg,肺血管肺血管抵抗≧3wood単位への基準改定が提案され話題となったが2),小児でも同基準が採用されることとなった3).肺動脈圧上昇は,閉塞性血管リモデリング,肺血管の収縮,先天性左右短絡,左心系疾患による左心房圧の上昇,呼吸器疾患,肺静脈病変など多岐にわたる機序によりもたらされる.WHO臨床分類では,これらの機序により大きく5群に分けられている1).小児でも,PHと関連する肺血管病変は,特発性や遺伝子異常,心疾患,呼吸疾患や様々な全身疾患を基盤に発症するが4),WHO臨床分類は,小児のPHの実地臨床には適用が難しい場合もあり,小児に特化した分類も提唱されている(いわゆるPanama分類)5)(Table 1).Panama分類では,周産期の適応障害,肺の発達成長障害,および肺低形成の重要性が強調されており6),カテゴリー1, 2には,詳細な周産期や発達の障害が挙げられ,独立したカテゴリー4として気管支肺異形成(BPD)が挙げられている(Table 1).カテゴリー1は,1.1母体または胎盤異常,1.2胎児の肺血管発達異常,1.3胎児の心疾患に分けられ,先天性横隔膜ヘルニア(CDH)は,カテゴリー1.2.1(Associated with fetal pulmonary hypoplasia)の「1.2.1.c」と分類される.
Table 1 10 Basic Categories of Pediatric Pulmonary Hypertensive Vascular Disease*5)| Category | Description |
|---|
| 1 | Prenatal or developmental pulmonary hypertensive vascular disease |
| 2 | Perinatal pulmonary vascular maladaptation |
| 3 | Pediatric cardiovascular disease |
| 4 | Bronchopulmonary dysplasia |
| 5 | Isolated pediatric pulmonary hypertensive vascular disease (isolated pediatric PAH) |
| 6 | Multifactorial pulmonary hypertensive vascular disease in congenital malformation syndromes |
| 7 | Pediatric lung disease |
| 8 | Pediatric thromboembolic disease |
| 9 | Pediatric hypobaric hypoxic exposure |
| 10 | Pediatric pulmonary vascular disease associated with other system disorders |
| *pulmonary hypertensive vascular diseaseと記載され肺高血圧(pulmonary hypertension)という記述ではない.厳密な肺高血圧の定義に当てはまらない病態も含めて,pulmonary hypertensive vascular diseaseと記載されている. |
小児PHの特徴あるいは課題の一つに,小児における治療効果などのエビデンスが少ないことが挙げられるが,小児の肺動脈性肺高血圧(PAH)に対しては,成人領域での知見に基づくアルゴリズムが適用され一定の効果を示している.一方で,肺発達成長障害や周産期の障害に関連する病態(BPDやCDH)を伴う慢性的なPHに対する治療に関する知見は乏しい.PAH治療薬開発や病変形成機序開発を目指した細胞・分子生物学的研究は,PAHに見られる遺伝子異常,血管病変や血管機能変化,PAHの動物モデルを中心に進められてきが,BPDやCDHに見られる病変を再現する動物モデルを用いた研究からも新たな標的分子やシグナルが同定されている.本稿では,肺の発達の機序,PAHの病態,モデル動物の解説に続けて,小児期の肺発達に関連する病態と関連する動物モデルについて概説する.
BPDは,胎生22週から28週の,未熟な肺への障害に起因し,CDHでは,横隔膜の形成される胎生10週以降,肺は物理的圧迫による障害を受ける可能性がある.
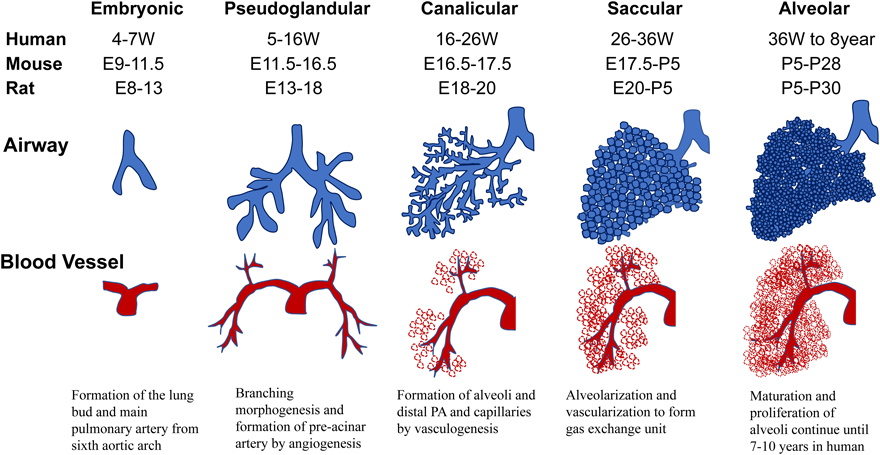
肺の形成は,胎生4週に前腸(anterior foregut)の腹側腹側壁から肺芽(lung bud)が膨らむように発生することから始まる.血管は第6大動脈弓から肺芽に向かい,血管が分岐し主肺動脈の起源となる(Fig. 1).横隔膜は胎生4週から10週で形成される.近位の気道(気管支,細気管支,終末細気管支)と肺動脈(pre-acinar artery)は胎生16週までにその分岐の大部分を完了し(branching morphogenesis),遠位の気道(呼吸細気管支,肺胞管,肺胞)と血管は,胎生16週以降に形成される.近位の血管は主肺動脈から分岐してangiogenesisにより伸展し,末梢血管(intra-acinar artery)と毛細血管は,胎生16週頃から血管構造のない間質にvasculogenesisにより形成される.その後,末梢肺動脈,毛細管,肺胞は,互いに密着し,胎生26週頃には原始肺胞が形成されガス交換が可能となる7–9).肺胞は胎生36週頃には成熟肺胞となり,3歳ごろまでは,急速にその数を増やす.3歳以降も,肺胞増加は続くが増加は緩徐となる10).マウス,ラットなどのげっ歯類は,ヒトに比べ未熟な肺発達段階で出生することは実験動物モデルでは重要である.ヒトの出生時の肺胞数は20×106個であるが,その8歳頃には成人と同様(300×106個)となると推定されている11, 12).pre-acinar arteryは生下時には,厚い壁を持つが,生後4か月頃には,末梢の肺血管の拡張により,成人と同様レベルまで薄くなる10).
肺形成過程の分子機構は不明な点も多いが,いくつかの中心的な役割を果たす分子が示され,様々な肺疾患の理解に新たな展開をもたらしている13).肺の発生初期,肺芽では転写因子Nkx2.1が発現し,近位の気道形成にはSox2の発現が重要である.branching morphogenesisの過程には,FGF10, Fgfr2, BMP4, sonic hedgehogなどが関与し,末梢の気道の形成にはSox9, Id2などの働きが重要と考えられている.肺を形成する細胞種は,上皮細胞,内皮細胞(血管,リンパ管),胸膜細胞,平滑筋細胞(気道,血管),周皮細胞,繊維芽細胞,ニューロン,免疫細胞(肺胞マクロファージ)など多彩であり,これらの細胞の起源について様々な議論がある14).
PAHの病態や病変の記述には,BPDやCDHなどPH全体に共通する要素が含まれるため,ヒトPAHの病理とPAH実験動物モデルの病変に関して基本的な事項をまず概説する.
肺動脈性肺高血圧の病因
PAHの病因面では,2000年に骨形成因子(bone morphogenetic protein, BMP)受容体II型遺伝子(BMPR2)が同定された15).In vitroでのBMPR2の機能については,血管内皮細胞では生存因子として16),血管平滑筋細胞では増殖抑制因子として働き17, 18),炎症細胞浸潤などの機序を通して血管炎症制御にも関わることが示されている19, 20).遺伝的背景の理解も進み,BMPR2遺伝子異常保持者のうちPAHを発症するのは約20%と低い浸透率を示すことから21),PAH発症には遺伝子異常と環境因子の連関が推定され,BMPR2は疾患感受性遺伝子として理解されている.
肺動脈性肺高血圧の病変形成と機序に関する課題
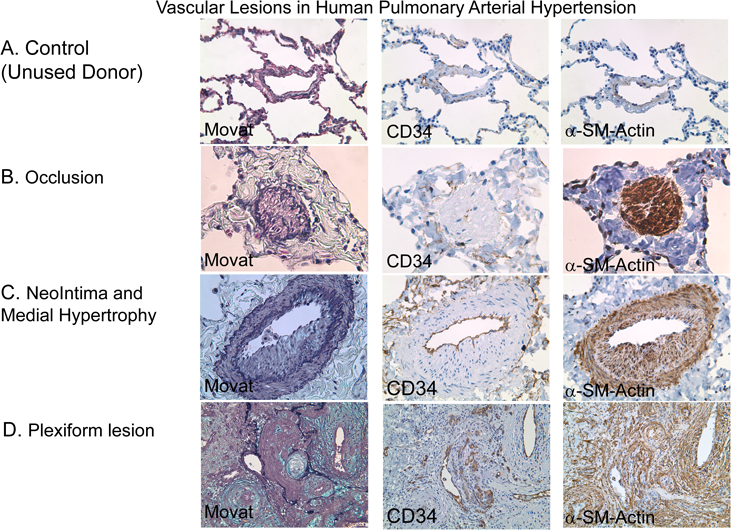
PAHは病因的にはheterogeneousな疾患であるが,肺血管病変として共通する部分は以下に集約され,これらは,BPDやCDHの血管病変と共通する部分もある.①血管拡張因子の低下22),収縮因子の上昇23),血管平滑筋の機能変化24)による血管収縮,②末梢血管の減少25),③遠位末梢動脈の筋性化,④血管平滑筋増殖による中膜肥厚,⑤閉塞性新生内膜病変,⑥叢状病変,⑦炎症細胞浸潤である.肺移植を実施されたPAH患者から摘出された肺組織に見られた,肺血管病変を示す20)(Fig. 2).閉塞した末梢肺動脈(Fig. 2B)の内腔では,α-smooth muscle actin(SMA)を発現する細胞を認める.また,肥厚した内膜病変(Fig. 2C)にもα-SMA陽性細胞が存在する.叢状病変(plexiform lesion, Fig. 2D)でもα-SMA陽性細胞を認め,壁の肥厚した病変血管周辺にはCD34陽性細胞の集積が見られ,異常な血管チャネル形成が示唆される.これらの病変のPAH病態における役割については,様々な議論が続けられている.①血管収縮と②~⑥の肺血管リモデリングの,いずれがどの程度本症の肺血管抵抗の上昇に寄与するのか26),また③遠位末梢動脈の筋性化や⑤新生内膜病変を担う細胞の起源は詳細には理解されていない.新生内膜病変については,血管内皮細胞の異常増殖とする報告が見られるが27),内膜にはα-SMAを発現する細胞も見られ(Fig. 2),その由来に関しては,議論が分かれている28–30).叢状病変ではアポトーシス抵抗性内皮細胞のクローナルな増殖や血管内皮前駆細胞の関与が示されているが31–33),これらの機序には慎重な意見もある.また,BMPR2などの疾患関連遺伝子の変異の病変形成機序における役割は未解明である.
肺動脈性肺高血圧の動物モデル
PHの領域で利用されてきた“標準的”モデルとして,モノクロタリン(MCT),慢性低酸素暴露について示し,比較的最近開発された血管内皮増殖因子(VEGF)受容体阻害剤SU5416+慢性低酸素暴露(Sugen/hypoxia)モデル34, 35)について概説する.
a)モノクロタリンモデル
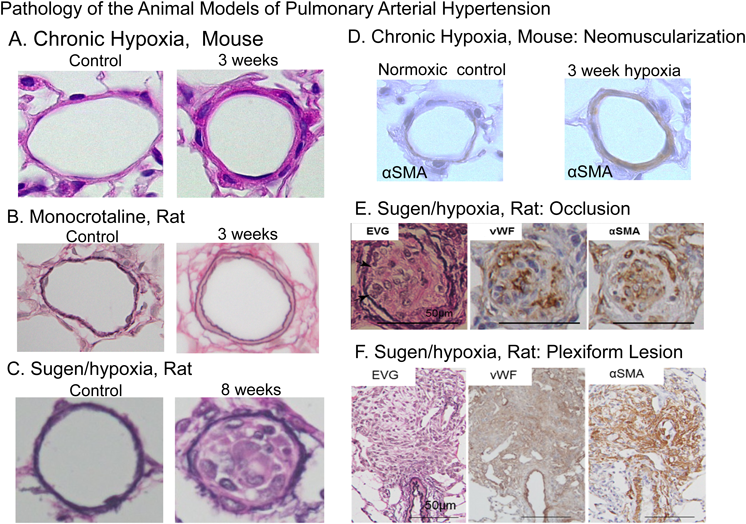
1960年代の研究で,ラットにマメ科植物Crotalaria spectabilisの種を,経口摂取させると,右室拡大を伴ううっ血性心不全で死亡することが知られていた36).その後,MCT(60 mg/kg)1回の皮下注射により,初期の血管障害に引き続き,14日後には右室肥大を示すことが示された37).MCT投与後,2~4日には,代謝産物monocrotaline pyrroleによる内皮障害を示し,MCT投与後8日目には,通常平滑筋層のない(normally non-muscular)末梢肺動脈への平滑筋層の出現(muscularization)が観察されるようになる(Fig. 3B).投与12日目には肺動脈圧上昇と右室肥大を認める37–40).MCT投与後8日目には,有意な肺への炎症細胞(マクロファージ)浸潤を認める41).血管病変とPHは進行し,投与後3週ごろから死亡する個体が現れ,5週以降の生存率は30%以下である42–44).MCTモデルでは,進行期でも内膜病変形成は認めない.MCTモデルにおいてもBMPR2の発現低下やBMPシグナリング減弱が示されており,ヒトPAHとの機序における接点を示唆する報告と考えられる45, 46).MCTの毒性には種差があり,マウスではMCT投与によりPHは誘発されない.
b)慢性低酸素暴露モデル
急性の低酸素下で肺動脈圧が上昇することはBradfordとDeanにより最初の記載があり47),実験的にはVon EulerとLiljestrandにより観察され48),心臓カテーテル法の導入後,ヒトでも同様の現象が観察された49).ラットやマウスの慢性低酸素暴露では,チャンバー内の10%酸素濃度環境が用いられることが多い.窒素ガス混入により酸素濃度をコントロールする(normobaric)またはチャンバー内から持続的に脱気を行うことによりチャンバー内の気圧を1/2気圧にコントロールする(hypobaric)装置が使用されている.本モデルでは,低酸素環境への暴露により,初期には急性の低酸素性肺血管収縮が惹起され,低酸素暴露4日目には慢性の肺動脈圧上昇が観察される50).その後,2週間にかけて肺動脈圧は上昇を続ける.
c)SU5416投与+慢性低酸素暴露モデル
MCTモデルや慢性低酸素モデルでの,肺血管の構造変化は,末梢の通常平滑筋層を認めない血管へのα-SMA陽性細胞層の出現(muscularization)であり(Fig. 3A, B, D),ヒトPAH(Fig. 2)のように新生内膜の形成は認めない50).これは,PAH研究を行う上での大きな課題であったが,2010年に内膜病変や叢状病変を認めるヒトPAHに類似するモデルとして登場したのがSU5416投与+慢性低酸素暴露(Sugen/hypoxia)モデルである35).
Sugen/hypoxiaモデルを開発したグループの研究者は,『ヒトPAHの病変血管では,異常な血管内皮細胞の増殖による不規則な血管新生が見られる』という観察に基づき,VEGFの阻害によるPAHモデルの作成の着想を得たと述べている51).仮説は,VEGFは血管内皮細胞の維持に重要な因子であり,VEGFの阻害により血管内皮細胞は機能障害から細胞死に至り,アポトーシス抵抗性の血管内皮細胞の異常増殖が誘発され,PAH病変を形成するという機序であった.VEGF受容体阻害剤SU5416は,Sugen社において,がん治療薬として開発されたVEGF受容体/tyrosine kinaseを阻害する低分子化合物であり,high-throughput screeningの過程で発見された52).SU5416の血管新生阻害は,他の実験動物においても実証されている53, 54).正常酸素下でのSU5416の投与では,軽度の肺動脈圧の上昇を認めるのみであった.慢性低酸素暴露と組み合わせたモデルは,SU5416(20 mg/kg)の単回皮下投与後,低酸素環境に3週間暴露することにより,肺小動脈に内皮細胞の増殖による閉塞性病変を認めるモデルとして報告された34).通常,ラットを3週間の低酸素暴露後に正常酸素環境に戻して飼育すると,2~3週後にはPHは軽快し,肺血管病変も退縮するが,Sugen/hypoxiaモデルでは3週間の低酸素暴露の後,正常酸素環境に戻した後もPHと血管病変は進行し,組織的には,叢状病変を認めるようになることが示された35, 55, 56)(Fig. 3C, E, F).このSU5416の単回皮下投与/低酸素暴露/正常酸素再開によるモデルが,現在Sugen/hypoxiaモデルとして,病変形成機序や薬効評価の研究で用いられるようになった51).同モデルを用い,エンドセリンレセプター拮抗薬による病変退縮効果を検証したところ,後期に見られる線維性閉塞病変は治療抵抗性を示し,早期治療の有益性を示す実験結果を示した57).Sugen/hypoxiaモデルは,マウスにおいても作成が試みられ,高度PH,内膜形成を伴う血管リモデリングを認めると報告されている58, 59)が,内膜形成による血管閉塞性病変は認められるが,稀であり,高度PHには寄与しないという見解や,マウスでのSugen/hypoxiaモデルの再現は困難であるとする意見も見られる60).
d)遺伝子改変モデル
PAHの病因に関連が示唆される様々な遺伝子をターゲットとし,全組織での遺伝子改変に加え,組織特異的,時間特異的に遺伝子発現を制御し肺血管病変形成などの表現型が解析されている.遺伝性PAH感受性遺伝子BMPR2の発見以降は,BMPR2遺伝子改変モデルの解析が進んでいるが課題も多い.誌面の制限のため,遺伝子改変モデルについては,記述に替え,Gomez-Arroyoらの総説60)を挙げる.
気管支肺異形成の病態
BPDは,28週以下の早産児(肺の発生段階ではcanalicular期,Fig. 1)への機械的刺激や酸素,炎症など様々な障害により発生する疾患であり,急性呼吸障害を発症し,人工呼吸器により酸素を投与された早産児においては,感染や炎症,酸素毒性,人工呼吸による物理的刺激(baro- or volu-trauma)が生後の肺発達や成熟を阻害することによるとされる61).Northwayらにより最初に記載された62)酸素毒性や肺の過伸展を主な機序として捉えるBPDの概念をold BPDと呼ぶのに対して,最近の極めて未熟な早産児に見られるBPDは,“new BPD”と呼ばれ,肺発達の阻害という機序に重点が置かれる63).本病態について,日本国内では1990年代から臨床症状,X線所見(泡沫状気腫状陰影),子宮内炎症(絨毛膜羊膜炎や臍帯炎など)などにより,元来のBPDを含む,新生児慢性肺疾患(CLD)を定義分類されてきたことから,CLDの疾患名が主に使用されている64).欧米では2000年のNational Institute of Child Health and Human Development(NICHD)のworkshopで新しい“BPD”として定義65)され,現在欧米を中心とする論文などではBPDという疾患名が主に用いられている.このような国内外での定義の差異には留意が必要であるが,本稿で引用する多くの文献では,NICHDの基準を採用していることから,本稿ではBPDの記載とする.
BPDに伴う肺の変化は,遠隔期予後にも影響を与え,学童期から成人期にもPHが持続することが最近示され66, 67),成育サイクルを通しての管理への議論が必要である.最近の周産期管理の改善により,超早産児の生存率の上昇により,BPDの罹患率は増加すると考えられ,BPDに伴う肺血管障害に対して,小児循環器医にも深い理解が求められる68).
気管支肺異形成の病理
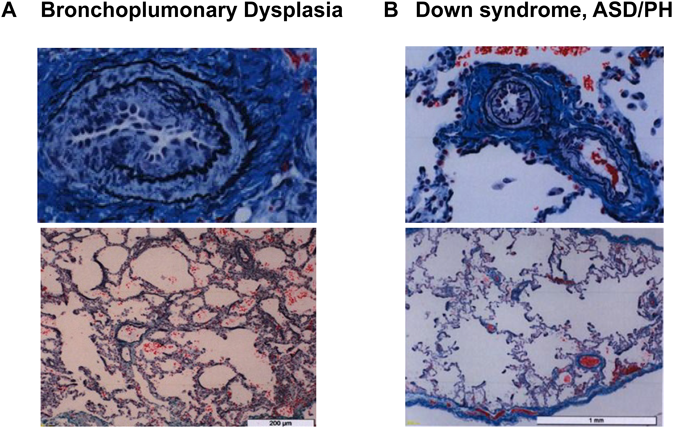
BPDに見られる肺組織の変化はでは,肺胞の形成不全と肺血管の発達成長障害により特徴づけられる6, 63).肺発達段階の阻害により,肺胞は大きくなり数は減少し(alveolar simplification),結果的にガス交換面積が減少する69).肺胞壁の肥厚により,肺胞内の吸入気と肺胞毛細管のガス拡散距離が拡大する(Fig. 4A).肺胞形成異常は,ダウン症候群などの染色体異常児の肺でも見られる(Fig. 4B).このような肺構造の変化は,適切なガス交換を障害しPHなどの肺血管障害の原因となる70).また新生児期早期のPHは重症BPD発症の予測因子であること71)も示されており,BPDの病態には血管と気道相互の影響が重要であることが示唆される.
気管支肺異形成の動物モデル
BPDの動物モデルには,マウス72),ラット73)などのげっ歯類が広く用いられるが,ウサギ74),未熟ヒツジ75),未熟ブタ76),ヒヒ77)などの大型実験動物も用いられる.マウスとラットが主に用いられる理由として,妊娠期間が短いこと,生後2~3週間で肺の成熟を完了する(Fig. 1)こと,そして満期で出生した時,肺の発達段階は,saccular stageであり(Fig. 1),ヒトの在胎26週程度で出生するヒトと同等の発達段階であることが挙げられる.これらは満期で出生したマウス,ラットを早産の肺障害モデルと用いることの妥当性を支持するが,マウス,ラットは,saccular stageの肺で必要なガス交換が可能であり,この点はヒトの病態と厳密には異なる点は,注意が必要である72, 73, 78, 79).動物モデルでBPDを作成するために,a)高濃度酸素投与(hyperoxia),b)出生前の炎症により肺病変を惹起するモデルc)VEGFシグナル阻害d)人工換気による肺進展などが使用されている.
a)高濃度酸素投与モデル
新生ラットを7~14日,高濃度酸素(60~100%)に曝露するモデルを用いBPDの病態が検討されている73).新生ラットに95%の高濃度酸素を投与したモデルでは,肺胞の拡大,肺胞数の減少,肺血管密度の減少などヒトBPDと同様の肺病変を認め,PHが惹起され80),同モデルでは,血管内皮依存性の血管弛緩反応が低下している81).新生ラットの7日間の高濃度酸素暴露は,10か月齢の成体ラットの肺胞構造にも影響することが示されており82),周産期の障害が成人期に与える影響が動物モデルでも示唆される.
b)出生前炎症モデル
絨毛羊膜炎(chorioamnionitis)など胎内での炎症はヒトBPDの増悪因子である.出生前にエンドトキシンを羊水内に投与すると,高濃度酸素BPDモデルの肺気道病変を増悪させ83),肺血管病変を増悪させることが示されている84).
c)血管内皮増殖因子シグナル阻害
上記のBPDモデルでは,VEGFシグナリング低下と肺血管の減少が示されているが,SU5416投与により新生ラットのVEGFシグナルを阻害し,血管新生を抑制すると,肺胞形成も阻害され,BPDと同様の肺所見が形成される53, 85).VEGFシグナリングの阻害因子である,soluble fms–tyrosine kinase1(Flt1)を妊娠ラットの羊腔内に投与すると,新生ラットにBPD同様の変化をもたらす86).この所見は,臨床的にFlt1はpreeclampsiaの羊水で上昇し,preeclampsiaはBPD発症のリスク因子であることに一致する87–89).これらの所見は,気道の形成と血管形成が,相互に影響することを示し,BPDにおける肺発達成長障害とPH発症の関係を考える上で重要である.
d)人工呼吸器を用いた肺過伸展モデル
未熟ヒヒ90),未熟ヒツジなど91)の大型実験動物を1~3週間人工呼吸管理を行うと,ヒトBPDと同様の肺の組織変化をきたす.これらのモデルは,未熟動物を使用していることから,“new BPD”をよく再現するモデルである.これらのモデルは,高頻度振動換気92)や一酸化窒素吸入療法93)など,主に治療介入の効果検証目的で利用されている.
気管支肺異形成動物モデルを用いた肺高血圧治療
治療効果としては,sildenafilなどcyclic GMP系薬剤の効果がラットモデルに用いられている.sildenafilは,hyperoxia BPD-PHモデルにおいて,PHと肺胞発育障害を改善することが示されている80).グアニル酸シクラーゼ刺激剤であるriociguatもラットにおいて,肺胞構造変化の改善に伴いPHが改善されることが示されている94).マウスのBPDモデルでは,PDE5の増加とcyclic GMPの減少が示され,sildenafilによりこれらの回復とともにPHが改善することが示されている95).また,PDE5阻害により血管新生が促進されることがin vitroでも示され96),sildenafilによりBPD肺の血管新生促進の可能性も示されている.これらの一連の研究成果が,sildenafilが臨床的BPD–PHにしばしば使用されている理由と考えられるが97),その効果や機序について臨床的に高いエビデンスを示す研究はなく今後の課題である.
先天性横隔膜ヘルニアの病態理解
CDHは,比較的頻度の高い(3000出生に1)疾患であり,先天的な横隔膜の欠損により,胎内で,腹部臓器が胸腔内に脱出し,心臓肺などを圧迫し,出生後の新生児に,新生児遷延性肺高血圧を伴う高度の低酸素血症をきたす,予後不良疾患である98).高度の肺低形成,左心室低形成,左心室機能低下,他の染色体異常や奇形症候群の合併などが,予後不良因子である.PHは,新生児期の手術後も持続する場合があり,亜急性期,遠隔期まで持続し,死亡率上昇と関連することが報告されている99).
先天性横隔膜ヘルニアの病理
CDHの肺では,肺胞増加の阻害,肺胞壁の肥厚などの気道の異常,肺血管の密度の減少,外膜と中膜の肥厚が見られ,血管病変や通常筋層を持たないintra-acinar arteryにも筋性化が見られる100–103).
胸腔内臓器の圧迫をきたす同様の新生児疾患であるcongenital pulmonary airway malformation(CPAM)に見られる肺低形成やPHは,CDHに見られるPHよりも軽度であることから104),CDHの重症PH発症には胸腔内臓器の圧迫以外の因子が関与すると考えられている.横隔膜欠損を生じる遺伝子欠損マウスが多数報告されているが,それらの多くが,肺の形成異常も伴うことはこの仮説を支持し,臨床的にもいくつかのCDH原因遺伝子が同定され,横隔膜ヘルニアに伴うPH発症には遺伝子異常や環境因子の関与の程度が大きいとされている105).染色体異常や奇形症候群に伴うcomplex CDHが40%程度あることや,染色体の部分欠失などの解析からも106),CDH発症における遺伝子異常の役割が推定されている.臨床的にはCDHの家族内の検索から,ZFPM2(FOG2)107, 108),GATA4109) and GATA6110)などの遺伝子が疾患遺伝子として挙げられている.
先天性横隔膜ヘルニアのモデル
CDHの病態研究には,げっ歯類では,a)teratogenic,b)geneticが用いられ,ヒツジなど大動物では,c)胎児期手術により作成したモデルが用いられている.外科的モデルはウサギでも作成される111).
a)teratogenicモデル
もっともよく使われるモデルは,nitrofenモデルである.げっ歯類の母体にnitrofen投与を行った場合,生後間もなく呼吸不全とチアノーゼを来たし死亡することが知られていた112).nitorofenの催奇形性は器官形成期に生じるため,nitrofen(50 mg/kg/day)を,ヒトの胎生4~6週に相当する,E8-9(マウス),E8-12(ラット)の妊娠母個体に経口投与すると,ヒトCDH類似の横隔膜の欠損と肺低形成を生じる.興味深いことに,投与時期により,生じるCDHの左右が異なる.早期の投与により左側のCDHを生じるのに対し,後期の投与では,右側CDHとなる113).
b)遺伝子異常モデル
臨床的CDHで見られた単一遺伝子異常の数が限られるのに対し,遺伝子改変マウスでは,現在までに,70種類以上の遺伝子欠損により,その表現型の一部として横隔膜欠損を生じることが知られている105, 106).retinoid signaling pathway114)やCOUP-TFII(NR2F2)115),FOG2108),GATA4116),WT1117),PBX1118)などが知られている.これらの遺伝子欠損マウスでは,横隔膜欠損と同時に,肺の形成異常も伴う.転写因子PBX1の欠損により,横隔膜欠損を生じることが示されているが,同マウスでは,血管収縮弛緩因子のアンバランスをきたし,新生児期の肺血管拡張が阻害され,PHに関わっていることが示された118).横隔膜形成と肺循環を同時に制御するシグナルとして興味深いモデルである.
c)ヒツジ胎仔手術モデル
妊娠羊に,妊娠80~110日で胎児に横隔膜欠損を作成する119).げっ歯類のモデルが,CDHの発症機序や遺伝的側面の理解に利用されるのに対し,大型動物のモデルはCDHの治療介入の研究につながり,本モデルを用い,気管閉塞により肺の発育が促されることを示す一連の研究から120),臨床での,fetal endoscopic tracheal occlusionの臨床試験につながった121).
先天性横隔膜ヘルニア動物モデルへの治療効果
nitorofenモデルの母体ラットにsildenafilを投与すると,CDHを伴う新生ラットの肺病変とPHが改善すると報告されている122).またsildenafil+bosentanの投与よる効果も示されている123).nitrofenモデルや遺伝子改変モデルの多くが,出生後早期に致死であり生体での評価が困難であり,胎児期の肺に対する母胎投与の治療効果を示すことに限られていた.最近報告された研究では,全身ノックアウトでは横隔膜欠損を生じて致死である転写因子PBX1の遺伝子を,肺組織のみで欠損させたコンディショナルノックアウトを作成し,生存可能なモデルを用い横隔膜形成に関わる遺伝子が生後早期の肺循環異常をきたすことを示している118).PBX1欠損マウスでは肺血管拡張薬に抵抗性を示すようである.臨床的に今後,横隔膜欠損と肺循環障害の新たな理解につながることが期待される.
引用文献References
1) Simonneau G, Gatzoulis MA, Adatia I, et al: Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62 Suppl: D34–D41
2) Simonneau G, Montani D, Celermajer DS, et al: Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53: 1801913
3) Rosenzweig EB, Abman SH, Adatia I, et al: Paediatric pulmonary arterial hypertension: Updates on definition, classification, diagnostics and management. Eur Respir J 2019; 53: 1801916
4) Abman SH, Hansmann G, Archer SL, et al: Pediatric Pulmonary Hypertension: Guidelines from the American Heart Association and American Thoracic Society. Circulation 2015; 132: 2037–2099
5) del Cerro MJ, Abman S, Diaz G, et al: A consensus approach to the classification of pediatric pulmonary hypertensive vascular disease: Report from the PVRI Pediatric Taskforce, Panama 2011. Pulm Circ 2011; 1: 286–298
6) Abman SH, Baker C, Gien J, et al: The Robyn Barst Memorial Lecture: Differences between the fetal, newborn, and adult pulmonary circulations: Relevance for age-specific therapies (2013 Grover Conference series). Pulm Circ 2014; 4: 424–440
7) Hislop A, Reid L: Development of the acinus in the human lung. Thorax 1974; 29: 90–94
8) deMello DE, Sawyer D, Galvin N, et al: Early fetal development of lung vasculature. Am J Respir Cell Mol Biol 1997; 16: 568–581
9) deMello DE, Reid LM: Embryonic and early fetal development of human lung vasculature and its functional implications. Pediatr Dev Pathol 2000; 3: 439–449
10) Hislop A, Reid L: Pulmonary arterial development during childhood: Branching pattern and structure. Thorax 1973; 28: 129–135
11) Dunnill MS: Quantitative methods in the study of pulmonary pathology. Thorax 1962; 17: 320–328
12) Davies G, Reid L: Growth of the alveoli and pulmonary arteries in childhood. Thorax 1970; 25: 669–681
13) Herriges M, Morrisey EE: Lung development: Orchestrating the generation and regeneration of a complex organ. Development 2014; 141: 502–513
14) Nikolić MZ, Sun D, Rawlins EL: Human lung development: Recent progress and new challenges. Development 2018; 145: dev163485
15) Lane KB, Machado RD, Pauciulo MW, et al: International PPH Consortium: Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet 2000; 26: 81–84
16) Teichert-Kuliszewska K, Kutryk M, Kuliszewski M, et al: Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: Implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res 2006; 98: 209–217
17) Morrell NW, Yang X, Upton PD, et al: Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation 2001; 104: 790–795
18) Zhang S, Fantozzi I, Tigno DD, et al: Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2003; 285: L740–L754
19) Hagen M, Fagan K, Steudel W, et al: Interaction of interleukin-6 and the BMP pathway in pulmonary smooth muscle. Am J Physiol Lung Cell Mol Physiol 2007; 292: L1473–L1479
20) Sawada H, Saito T, Nickel NP, et al: Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J Exp Med 2014; 211: 263–280
21) Newman JH, Wheeler L, Lane K, et al: Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med 2001; 345: 319–324
22) Tuder RM, Cool CD, Geraci MW, et al: Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999; 159: 1925–1932
23) Giaid A, Yanagisawa M, Langleben D, et al: Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med 1993; 328: 1732–1739
24) Yuan JX, Aldinger AM, Juhaszova M, et al: Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 1998; 98: 1400–1406
25) Rabinovitch M, Haworth SG, Castaneda AR, et al: Lung biopsy in congenital heart disease: A morphometric approach to pulmonary vascular disease. Circulation 1978; 58: 1107–1122
26) Rabinovitch M, Chesler N, Molthen RC: Point:Counterpoint: Chronic hypoxia-induced pulmonary hypertension does/does not lead to loss of pulmonary vasculature. J Appl Physiol (1985) 2007; 103: 1449–1451
27) Tuder RM, Groves B, Badesch DB, et al: Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol 1994; 144: 275–285
28) Qiao L, Nishimura T, Shi L, et al: Endothelial fate mapping in mice with pulmonary hypertension. Circulation 2014; 129: 692–703
29) Arciniegas E, Frid MG, Douglas IS, et al: Perspectives on endothelial-to-mesenchymal transition: Potential contribution to vascular remodeling in chronic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2007; 293: L1–L8
30) Sheikh AQ, Misra A, Rosas IO, et al: Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci Transl Med 2015; 7: 308ra159
31) Masri FA, Xu W, Comhair SA, et al: Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2007; 293: L548–L554
32) Lee SD, Shroyer KR, Markham NE, et al: Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J Clin Invest 1998; 101: 927–934
33) Asosingh K, Aldred MA, Vasanji A, et al: Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am J Pathol 2008; 172: 615–627
34) Taraseviciene-Stewart L, Kasahara Y, Alger L, et al: Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 2001; 15: 427–438
35) Abe K, Toba M, Alzoubi A, et al: Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 2010; 121: 2747–2754
36) Kay JM, Harris P, Heath D: Pulmonary hypertension produced in rats by ingestion of Crotalaria spectabilis seeds. Thorax 1967; 22: 176–179
37) Hilliker KS, Bell TG, Roth RA: Pneumotoxicity and thrombocytopenia after single injection of monocrotaline. Am J Physiol 1982; 242: H573–H579
38) Rosenberg HC, Rabinovitch M: Endothelial injury and vascular reactivity in monocrotaline pulmonary hypertension. Am J Physiol 1988; 255: H1484–H1491
39) Roth RA, Reindel JF: Lung vascular injury from monocrotaline pyrrole, a putative hepatic metabolite. Adv Exp Med Biol 1991; 283: 477–487
40) Meyrick B, Gamble W, Reid L: Development of Crotalaria pulmonary hypertension: Hemodynamic and structural study. Am J Physiol 1980; 239: H692–H702
41) Sawada H, Mitani Y, Maruyama J, et al: A nuclear factor-kappaB inhibitor pyrrolidine dithiocarbamate ameliorates pulmonary hypertension in rats. Chest 2007; 132: 1265–1274
42) Jasmin JF, Lucas M, Cernacek P, et al: Effectiveness of a nonselective ET(A/B) and a selective ET(A) antagonist in rats with monocrotaline-induced pulmonary hypertension. Circulation 2001; 103: 314–318
43) Abe K, Shimokawa H, Morikawa K, et al: Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circ Res 2004; 94: 385–393
44) Gomez-Arroyo JG, Farkas L, Alhussaini AA, et al: The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol 2012; 302: L363–L369
45) Morty RE, Nejman B, Kwapiszewska G, et al: Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol 2007; 27: 1072–1078
46) Zakrzewicz A, Kouri FM, Nejman B, et al: The transforming growth factor-beta/Smad2,3 signalling axis is impaired in experimental pulmonary hypertension. Eur Respir J 2007; 29: 1094–1104
47) Bradford JR, Dean HP: The Pulmonary Circulation. J Physiol 1894; 16: 34–158, 25
48) Euler US, Liljestrand G: Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand 1946; 12: 301–320
49) Motley HL, Cournand A, Werko L, et al: The influence of short periods of induced acute anoxia upon pulmonary artery pressures in man. Am J Physiol 1947; 150: 315–320
50) Rabinovitch M, Gamble W, Nadas AS, et al: Rat pulmonary circulation after chronic hypoxia: Hemodynamic and structural features. Am J Physiol 1979; 236: H818–H827
51) Nicolls MR, Mizuno S, Taraseviciene-Stewart L, et al: New models of pulmonary hypertension based on VEGF receptor blockade-induced endothelial cell apoptosis. Pulm Circ 2012; 2: 434–442
52) Fong TA, Shawver LK, Sun L, et al: SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res 1999; 59: 99–106
53) Jakkula M, Le Cras TD, Gebb S, et al: Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol 2000; 279: L600–L607
54) Keskin U, Totan Y, Karadag R, et al: Inhibitory effects of SU5416, a selective vascular endothelial growth factor receptor tyrosine kinase inhibitor, on experimental corneal neovascularization. Ophthalmic Res 2012; 47: 13–18
55) Toba M, Alzoubi A, O’Neill KD, et al: Temporal hemodynamic and histological progression in Sugen5416/hypoxia/normoxia-exposed pulmonary arterial hypertensive rats. Am J Physiol Heart Circ Physiol 2014; 306: H243–H250
56) Otsuki S, Sawada H, Yodoya N, et al: Potential contribution of phenotypically modulated smooth muscle cells and related inflammation in the development of experimental obstructive pulmonary vasculopathy in rats. PLoS One 2015; 10: e0118655
57) Shinohara T, Sawada H, Otsuki S, et al: Macitentan reverses early obstructive pulmonary vasculopathy in rats: Early intervention in overcoming the survivin-mediated resistance to apoptosis. Am J Physiol Lung Cell Mol Physiol 2015; 308: L523–L538
58) Ciuclan L, Bonneau O, Hussey M, et al: A novel murine model of severe pulmonary arterial hypertension. Am J Respir Crit Care Med 2011; 184: 1171–1182
59) Suzuki T, Carrier EJ, Talati MH, et al: Isolation and characterization of endothelial-to-mesenchymal transition-cells in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2018; 314: L118–L126
60) Gomez-Arroyo J, Saleem SJ, Mizuno S, et al: A brief overview of mouse models of pulmonary arterial hypertension: Problems and prospects. Am J Physiol Lung Cell Mol Physiol 2012; 302: L977–L991
61) Jain D, Bancalari E: Bronchopulmonary dysplasia: Clinical perspective. Birth Defects Res A Clin Mol Teratol 2014; 100: 134–144
62) Northway WH Jr., Rosan RC, Porter DY: Pulmonary disease following respirator therapy of hyaline-membrane disease: Bronchopulmonary dysplasia. N Engl J Med 1967; 276: 357–368
63) Coalson JJ: Pathology of new bronchopulmonary dysplasia. Paper presented at Seminars in neonatology 2003
64) 藤村正哲,田村正徳,森 臨太郎:改訂2版—科学的根拠に基づいた新生児慢性肺疾患の診療指針—.大阪,メディカ出版,2010
65) Jobe AH, Bancalari E: Bronchopulmonary dysplasia. Am J Respir Crit Care Med 2001; 163: 1723–1729
66) Zivanovic S, Pushparajah K, Calvert S, et al: Pulmonary artery pressures in school-age children born prematurely. J Pediatr 2017; 191: 42–49.e3
67) Goss KN, Beshish AG, Barton GP, et al: Early pulmonary vascular disease in young adults born preterm. Am J Respir Crit Care Med 2018; 198: 1549
68) Krishnan U, Feinstein JA, Adatia I, et al: Evaluation and management of pulmonary hypertension in children with bronchopulmonary dysplasia. J Pediatr 2017; 188: 24–34.e1
69) Day CL, Ryan RM: Bronchopulmonary dysplasia: Old becomes new again! Pediatr Res 2017; 81: 210–213
70) Baker CD, Abman SH: Impaired pulmonary vascular development in bronchopulmonary dysplasia. Neonatology 2015; 107: 344–351
71) Mourani PM, Sontag MK, Younoszai A, et al: Early pulmonary vascular disease in preterm infants at risk for bronchopulmonary dysplasia. Am J Respir Crit Care Med 2015; 191: 87–95
72) Berger J, Bhandari V: Animal models of bronchopulmonary dysplasia: The term mouse models. Am J Physiol Lung Cell Mol Physiol 2014; 307: L936–L947
73) O’Reilly M, Thébaud B: Animal models of bronchopulmonary dysplasia: The term rat models. Am J Physiol Lung Cell Mol Physiol 2014; 307: L948–L958
74) D’Angio CT, Ryan RM: Animal models of bronchopulmonary dysplasia: The preterm and term rabbit models. Am J Physiol Lung Cell Mol Physiol 2014; 307: L959–L969
75) Albertine KH: Utility of large-animal models of BPD: Chronically ventilated preterm lambs. Am J Physiol Lung Cell Mol Physiol 2015; 308: L983–L1001
76) Arrindell EL Jr., Krishnan R, Van Der Merwe M, et al: Lung volume recruitment in a preterm pig model of lung immaturity. Am J Physiol Lung Cell Mol Physiol 2015; 309: L1088–L1092
77) Yoder BA, Coalson JJ: Animal models of bronchopulmonary dysplasia. The preterm baboon models. Am J Physiol Lung Cell Mol Physiol 2014; 307: L970–L977
78) Hilgendorff A, Reiss I, Ehrhardt H, et al: Chronic lung disease in the preterm infant: Lessons learned from animal models. Am J Respir Cell Mol Biol 2014; 50: 233–245
79) Nardiello C, Mižíková I, Morty RE: Looking ahead: Where to next for animal models of bronchopulmonary dysplasia? Cell Tissue Res 2017; 367: 457–468
80) Ladha F, Bonnet S, Eaton F, et al: Sildenafil improves alveolar growth and pulmonary hypertension in hyperoxia-induced lung injury. Am J Respir Crit Care Med 2005; 172: 750–756
81) Dumas de la Roque E, Smeralda G, Quignard J-F, et al: Altered vasoreactivity in neonatal rats with pulmonary hypertension associated with bronchopulmonary dysplasia: Implication of both eNOS phosphorylation and calcium signaling. PLoS One 2017; 12: e0173044
82) O’Reilly M, Harding R, Sozo F: Altered small airways in aged mice following neonatal exposure to hyperoxic gas. Neonatology 2014; 105: 39–45
83) Choi CW, Kim BI, Hong J-S, et al: Bronchopulmonary dysplasia in a rat model induced by intra-amniotic inflammation and postnatal hyperoxia: Morphometric aspects. Pediatr Res 2009; 65: 323–327
84) Tang J-R, Seedorf GJ, Muehlethaler V, et al: Moderate postnatal hyperoxia accelerates lung growth and attenuates pulmonary hypertension in infant rats after exposure to intra-amniotic endotoxin. Am J Physiol Lung Cell Mol Physiol 2010; 299: L735–L748
85) Le Cras TD, Markham NE, Tuder RM, et al: Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol Lung Cell Mol Physiol 2002; 283: L555–L562
86) Tang J-R, Karumanchi SA, Seedorf G, et al: Excess soluble vascular endothelial growth factor receptor-1 in amniotic fluid impairs lung growth in rats: Linking preeclampsia with bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 2012; 302: L36–L46
87) Foidart J-M, Schaaps J-P, Chantraine F, et al: Dysregulation of anti-angiogenic agents (sFlt-1, PLGF, and sEndoglin) in preeclampsia: A step forward but not the definitive answer. J Reprod Immunol 2009; 82: 106–111
88) Hansen AR, Barnés CM, Folkman J, et al: Maternal preeclampsia predicts the development of bronchopulmonary dysplasia. J Pediatr 2010; 156: 532–536
89) Levine RJ, Lam C, Qian C, et al: Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med 2006; 355: 992–1005
90) Coalson JJ, Kuehl TJ, Escobedo MB, et al: A baboon model of bronchopulmonary dysplasia: II. Pathologic features. Exp Mol Pathol 1982; 37: 335–350
91) Albertine KH, Jones GP, Starcher BC, et al: Chronic lung injury in preterm lambs: Disordered respiratory tract development. Am J Respir Crit Care Med 1999; 159: 945–958
92) Kinsella JP, Gerstmann DR, Clark RH, et al: High-frequency oscillatory ventilation versus intermittent mandatory ventilation: Early hemodynamic effects in the premature baboon with hyaline membrane disease. Pediatr Res 1991; 29: 160–166
93) McCurnin DC, Pierce RA, Chang LY, et al: Inhaled NO improves early pulmonary function and modifies lung growth and elastin deposition in a baboon model of neonatal chronic lung disease. Am J Physiol Lung Cell Mol Physiol 2005; 288: L450–L459
94) Donda K, Zambrano R, Moon Y, et al: Riociguat prevents hyperoxia-induced lung injury and pulmonary hypertension in neonatal rats without effects on long bone growth. PLoS One 2018; 13: e0199927
95) Lee KJ, Berkelhamer SK, Kim GA, et al: Disrupted pulmonary artery cyclic guanosine monophosphate signaling in mice with hyperoxia-induced pulmonary hypertension. Am J Respir Cell Mol Biol 2014; 50: 369–378
96) Zhu B, Zhang L, Alexeyev M, et al: Type 5 phosphodiesterase expression is a critical determinant of the endothelial cell angiogenic phenotype. Am J Physiol Lung Cell Mol Physiol 2009; 296: L220–L228
97) Mourani PM, Sontag MK, Ivy DD, et al: Effects of long-term sildenafil treatment for pulmonary hypertension in infants with chronic lung disease. J Pediatr 2009; 154: 379–384, 384.e1–384.e2
98) Gien J, Kinsella J: Management of pulmonary hypertension in infants with congenital diaphragmatic hernia. J Perinatol 2016; 36 Suppl 2: S28–S31
99) Kinsella JP, Ivy DD, Abman SH: Pulmonary vasodilator therapy in congenital diaphragmatic hernia: Acute, late, and chronic pulmonary hypertension. Semin Perinatol 2005; 29: 123–128
100) Rottier R, Tibboel D: Fetal lung and diaphragm development in congenital diaphragmatic hernia. Paper presented at Seminars in perinatology 2005
101) Mohseni-Bod H, Bohn D: Pulmonary hypertension in congenital diaphragmatic hernia. Paper presented at Seminars in pediatric surgery 2007
102) Beals DA, Schloo BL, Vacanti JP, et al: Pulmonary growth and remodeling in infants with high-risk congenital diaphragmatic hernia. J Pediatr Surg 1992; 27: 997–1002
103) Sokol J, Bohn D, Lacro RV, et al: Fetal pulmonary artery diameters and their association with lung hypoplasia and postnatal outcome in congenital diaphragmatic hernia. Am J Obstet Gynecol 2002; 186: 1085–1090
104) Derderian SC, Jayme CM, Cheng LS, et al: Mass effect alone may not explain pulmonary vascular pathology in severe congenital diaphragmatic hernia. Fetal Diagn Ther 2016; 39: 117–124
105) Donahoe PK, Longoni M, High FA: Polygenic causes of congenital diaphragmatic hernia produce common lung pathologies. Am J Pathol 2016; 186: 2532–2543
106) Holder AM, Klaassens M, Tibboel D, et al: Genetic factors in congenital diaphragmatic hernia. Am J Hum Genet 2007; 80: 825–845
107) Longoni M, Russell M, High F, et al: Prevalence and penetrance of ZFPM2 mutations and deletions causing congenital diaphragmatic hernia. Clin Genet 2015; 87: 362–367
108) Ackerman KG, Herron BJ, Vargas SO, et al: Fog2 is required for normal diaphragm and lung development in mice and humans. PLoS Genet 2005; 1: 58–65
109) Yu L, Wynn J, Cheung YH, et al: Variants in GATA4 are a rare cause of familial and sporadic congenital diaphragmatic hernia. Hum Genet 2013; 132: 285–292
110) Yu L, Bennett JT, Wynn J, et al: Whole exome sequencing identifies de novo mutations in GATA6 associated with congenital diaphragmatic hernia. J Med Genet 2014; 51: 197–202
111) Chiu PPL: New insights into congenital diaphragmatic hernia–a surgeon’s introduction to CDH animal models. Front Pediatr 2014; 2: 36
112) Manson JM: Mechanism of nitrofen teratogenesis. Environ Health Perspect 1986; 70: 137–147
113) Greer JJ: Current concepts on the pathogenesis and etiology of congenital diaphragmatic hernia. Respir Physiol Neurobiol 2013; 189: 232–240
114) Greer JJ, Babiuk RP, Thebaud B: Etiology of congenital diaphragmatic hernia: The retinoid hypothesis. Pediatr Res 2003; 53: 726–730
115) You L-R, Takamoto N, Yu C-T, et al: Mouse lacking COUP-TFII as an animal model of Bochdalek-type congenital diaphragmatic hernia. Proc Natl Acad Sci USA 2005; 102: 16351–16356
116) Jay PY, Bielinska M, Erlich JM, et al: Impaired mesenchymal cell function in Gata4 mutant mice leads to diaphragmatic hernias and primary lung defects. Dev Biol 2007; 301: 602–614
117) Clugston RD, Klattig J, Englert C, et al: Teratogen-induced, dietary and genetic models of congenital diaphragmatic hernia share a common mechanism of pathogenesis. Am J Pathol 2006; 169: 1541–1549
118) McCulley DJ, Wienhold MD, Hines EA, et al: PBX transcription factors drive pulmonary vascular adaptation to birth. J Clin Invest 2018; 128: 655–667
119) De Lorimier A: Hypoplastic lungs in fetal lambs with surgically produced congenital diaphragmatic hernia. Surgery 1967; 62: 12–17
120) Luks FI, Wild YK, Piasecki GJ, et al: Short-term tracheal occlusion corrects pulmonary vascular anomalies in the fetal lamb with diaphragmatic hernia. Surgery 2000; 128: 266–272
121) Harrison MR, Keller RL, Hawgood SB, et al: A randomized trial of fetal endoscopic tracheal occlusion for severe fetal congenital diaphragmatic hernia. N Engl J Med 2003; 349: 1916–1924
122) Luong C, Rey-Perra J, Vadivel A, et al: Antenatal sildenafil treatment attenuates pulmonary hypertension in experimental congenital diaphragmatic hernia. Circulation 2011; 123: 2120–2131
123) Lemus-Varela Mde L, Soliz A, Gómez-Meda BC, et al: Antenatal use of bosentan and/or sildenafil attenuates pulmonary features in rats with congenital diaphragmatic hernia. World J Pediatr 2014; 10: 354–359