1. 概要
先天性QT延長症候群(long QT syndrome: LQTS)は心電図でのQT間隔の延長が特徴の遺伝性不整脈である.Torsade de pointes(TdP)と呼ばれる多形性心室頻拍(VT)や心室細動(VF)が惹起され,失神や突然死を引き起こす.心筋活動電位に関連したイオンチャネルをコードする遺伝子異常が主な原因である.現在までに少なくとも17の原因遺伝子が明らかになっている.
2. 疫学
頻度は,新生児を対象としたイタリアの研究では2,000人に1人1),日本の1か月健診での研究では約1,100人に1人と報告されている2).女性にやや多い傾向がある.学童期から思春期での発症が多く,形態異常を伴わない小児期院外心停止の原因としてはLQTSが最も頻度が高い3).
3. 病態生理
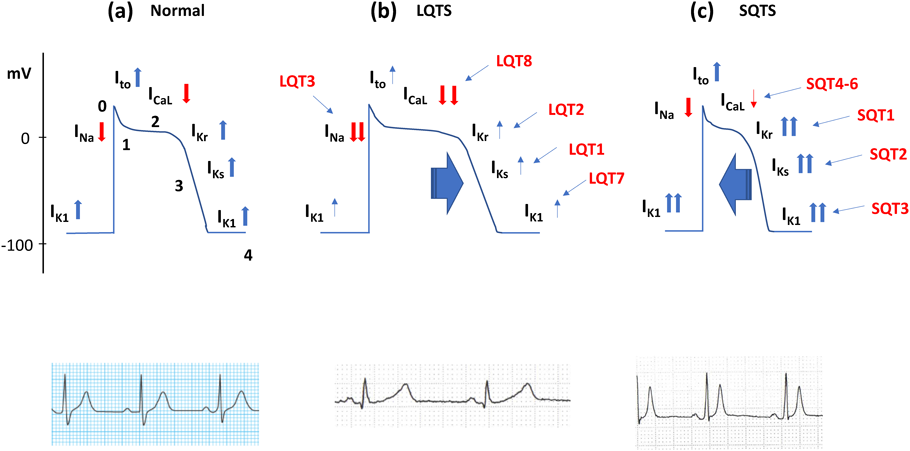
心室筋細胞の活動電位に関わる主なイオンチャネルには,Naチャネル,L型Caチャネルおよび複数のKチャネルがある.Na, L型Caチャネルでは細胞外から細胞内にそれぞれNa+, Ca2+が移動し,内向きの電流(INa,ICaL)が生じ,脱分極を起こすよう作用する.またKチャネルでは心筋細胞内から細胞外にK+が移動し,主に外向き電流が生じて再分極を促すよう作用する.Kチャネルは,脱分極早期に働く一過性外向きK電流(Ito)を生じるチャネルや脱分極後時間が経ってから働く遅延整流K電流(IKrやIKsなど)を生じるチャネル,さらに内向き整流性Kチャネル(IK1チャネル)がある(Fig. 1a). LQTSでは遺伝子異常によりNa, Caチャネルの機能亢進やKチャネルの機能低下などが起こり,再分極が遅れる.心室筋細胞レベルでは活動電位持続時間が延長し,心電図上ではQT間隔が延長する(Fig. 1b).早期後脱分極(EAD)からの突撃波による心室期外収縮が引き金となり,不均一な再分極に伴うphase 2 reentryがVTやVFを持続させる.
4. 遺伝的背景
遺伝子変異の報告以前より,先天性LQTSは先天性聾唖の有無で分類されていた.Jervell and Lange–Nielsen症候群は,先天性聾唖を伴い,稀で常染色体優性遺伝形式をとる.一方Romano–Ward症候群は先天性聾唖を伴わず,高頻度で常染色体劣性遺伝形式をとる.
その後遺伝子変異が次々と報告され(Table 1),現在では臨床診断されたLQTS患者の約75%に遺伝子変異が同定される.そのうちの90%以上を占めるLQT1-3では遺伝子型と表現型の関連が明らかになっている.さらに同じ遺伝子型でも変異部位や変異タイプによる表現型の違いも報告されている.主要な遺伝子型について以下の通り述べる.
Table 1 Genes causing long QT syndromes| Genetic Subtype | Disease-associated gene | Effect on channel function |
|---|
| Romano–Ward syndrome |
| LQT1 | KCNQ1 | ↓IKs |
| LQT2 | KCNH2 | ↓IKr |
| LQT3 | SCN5A | ↑INa |
| LQT4 | ANK2 | Multichannel interactions |
| LQT5 | KCNE1 | ↓IKs |
| LQT6 | KCNE2 | ↓IKr |
| LQT7 | KCNJ2 | ↓IK1 |
| LQT8 | CACNA1C | ↑ICa-L |
| LQT9 | CAV3 | ↑INa |
| LQT10 | SCN4B | ↑INa |
| LQT11 | AKAP9 | ↓IKs |
| LQT12 | SNTA1 | ↑INa |
| LQT13 | KCNJ5 | ↓IKACh |
| LQT14 | CALM1 | Multichannel interactions |
| LQT15 | CALM2 | Multichannel interactions |
| LQT16 | CALM3 | ↑ICa-L |
| LQT17 | TECREL | ↑ICa-L |
| Jervell and Lange–Nielsen syndrome |
| JNL1 | KCNQ1 | ↓IKs |
| JML2 | KCNE1 | ↓IKs |
| IKs, slow delayed rectifier potassium current; IKr, rapid delayed rectifier potassium current; INa, fast inward sodium current; IK1, background potassium current; ICa-L, L-type calcium channel current; Ito, early transient outward potassium current; IKACh, muscarinic-gated potassium current. |
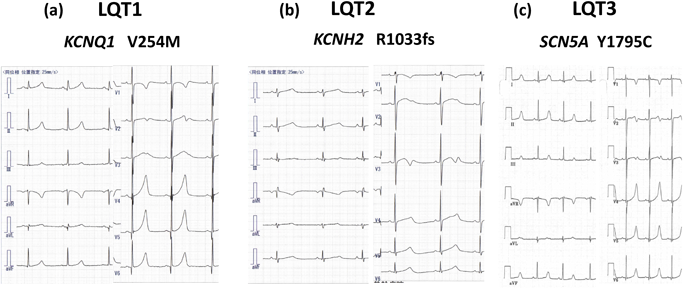
LQT1の原因遺伝子は,IKsを生じるチャネルをコードするKCNQ1である.LQT1ではチャネルの機能低下を生じる変異によりIKsが減少する.IKsは交感神経刺激により活性化されるため頻脈時の再分極の役割が大きい.よって運動時など頻脈時にQT間隔が延長し,心イベントを起こしやすい.
LQT2の原因遺伝子は,IKrを生じるチャネルをコードするKCNH2である.LQT2ではチャネルの機能低下を生じる変異によりIKrが減少する.IKrは急激な交感神経刺激を受けた際の再分極の役割が大きい.よって情動ストレスや目覚まし時計の音での覚醒などに心イベントを起こしやすい.
LQT3の原因遺伝子であるSCN5AはNaチャネルのαサブユニットをコードしている.INaは心筋活動電位の脱分極を起こし,速やかに不活性化する.LQT3ではチャネルの不活性化が障害されるため,再分極が遅れ,QT間隔が延長する.安静時,睡眠中に心イベントを起こしやすい.
LQT8はL型Caチャネルのα1サブユニットをコードする遺伝子CACNA1の異常による.L型Caチャネルの機能亢進により,ICa-Lが増加し,再分極が遅れ,QT延長が生じる.合指症など全身症状を伴うTimothy症候群として当初報告され,稀と考えられていた.しかし近年,全身症状を伴わないLQT8が報告され注目を集め,頻度は先天性LQTSの2.5%との報告もある4).LQT3に類似した遅発性T波を呈し,鑑別を要する5).
5. 診断
1)症状
典型的な症状は,失神,痙攣発作や突然死である.失神,痙攣は自然停止するTdPで引き起こされ,突然死の多くはTdPがVFに移行して起こると考えられている.一過性の意識障害や痙攣の鑑別診断としてLQTSを念頭におくべきである.
2)診断基準
日本循環器学会(日循)のガイドラインもHRS・EHRA・APHRS合同ステートメントの診断基準に従っている(Table 2)6, 7).同診断基準にあるLQTSリスクスコアはSchwartzスコア8)であり(Table 3),従来通り同スコアが3.5点以上のものはLQTSと確定診断される.さらに2次性QT延長の要因がないQTc≧500 msのQT延長,またはLQTSの遺伝子異常を認めるものもLQTSと確定診断する.
Table 2 Expert consensus recommendations on LQTS diagnosis7)| 1. LQTS is diagnosed; |
| a. In the presence of an LQTS risk score ≧3.5 in the absence of a secondary cause for QT prolongation and/or |
| b. In the presence of an unequivocally pathogenic mutation in one of the LQTS genes or |
| c. In the presence of a QT interval corrected for heart rate using Bazett’s formula (QTc) ≧500 ms in repeated 12-lead electrocardiogram (ECG) and in the absence of a secondary cause for QT prolongation. |
| 2. LQTS can be diagnosed in the presence of a QTc between 480 and 499 ms in repeated 12-lead ECGs in a patient with unexplained syncope in the absence of a secondary cause for QT prolongation and in the absence of a pathogenic mutation. |
Table 3 LQTS risk score8) | points |
|---|
| Electrocardiographic findings*1 | QTc*2 | ≧480 msec | 3 |
| 460~479 msec | 2 |
| 450~459 msec (male) | 1 |
| QTc*2 4th minute of recovery from exercise stress test | ≧480 msec | 1 |
| Torsade de pointes*3 | 2 |
| Visible T wave alternans | 1 |
| Notched T wave in 3 leads | 1 |
| Low heart rate for age*4 | 0.5 |
| Clinical history | Syncope*3 | With stress | 2 |
| Without stress | 1 |
| Congenital deafness | 0.5 |
| Family history*5 | A. Family members with definite LQTS | 1 |
| B. Unexplained sudden cardiac death younger than age 30 among immediate family members | 0.5 |
| Score: ≦1 point: low probability of LQTS; 1.5–3 points: intermediate probability of LQTS; ≧3.5 points: high probability. *1 In absence of medications or disorders known to affect these electrocardiographic features. *2 QTc calculated by Bazett formula where QTc=QT/√RR. *3 Mutually exclusive. *4 Resting heart rate below the second percentile for age. *5 The same family member cannot be counted in A and B. |
3)心電図診断
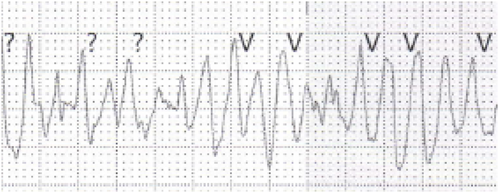
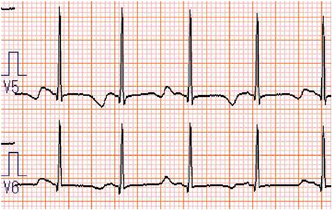
心電図で重要なのはQT延長,T波形,TdP, T波オルタナンス(TWA)の評価である.QT延長は,心拍数補正したQTcで評価する.確定診断にはBazett補正によるQTcを用いる.T波形は遺伝子型により特徴がある(Fig. 2). TdPは等電位線を軸にねじれるような波形変化をする特徴的なVTである(Fig. 3). TWAはT波の形態や極性が1拍ごとに変化する現象である(Fig. 4).心室再分極の不安定性を示し,認める例は心イベントのリスクが高い.
4)負荷試験・ホルター心電図
LQTSを疑う際は安静時心電図だけでは診断がつかないことがあるので必ずホルター心電図や運動負荷試験で評価する.LQT1の36%, LQT2の19%が安静時のQTcが正常または境界域と報告されている.小児の運動負荷はトレッドミルが一般的である.2011年に改定されたSchwartzのスコア8)(LQTSリスクスコア)でも運動回復期4分のQTc値が評価項目として追加された.さらに運動負荷によるQTcの変化は遺伝子型により特徴があり,遺伝子型の予測の参考にもなる9).ホルター心電図は運動負荷が困難な例にも行え,睡眠時の評価もできる利点がある.しかしフィルター設定次第でT波の終点が変化するので,ホルター心電図のQTcのみでの診断は慎重にすべきである10).
カテコラミン負荷試験も潜在性のLQTSの診断や遺伝子型の予測に有用である.運動負荷試験やホルター心電図での診断が困難な例には特に勧められる.しかしTdP誘発のリスクを伴うので患者への説明と十分な準備が必要である.プロプラノロールや硫酸マグネシウム等の緊急薬剤と除細動器を準備し,複数の医師の立ち会いのもとで行う.
顔面浸水負荷試験は自律神経活動変化に伴う心電図変化が評価できる.顔面浸水直後には交感神経優位で頻脈に,浸水を続けると副交感神経優位となり徐脈になる.小学校低学年以下では十分な施行が困難なことが多い.
5)遺伝子検査
遺伝子型による症状や有効治療の違いが明らかになり,LQTSが疑われる場合は,遺伝子検査が保険適応となった.遺伝子検査の最も良い適応は,LQTSと臨床診断された例の治療・管理方針の決定目的である.
6)学校心臓検診での注意点
小児では心拍数が高いためにBazett補正では過剰補正になる.そこで学校心臓検診など小児のスクリーニング時には,心拍数の影響がより少ないFridericia補正が推奨されている.自動計測法でQT延長として抽出した例は,QT間隔をマニュアル法(接線法)で再計測する.詳細は学校心臓検診のガイドライン11)を参照いただきたい.確定診断時はBazett補正を用いる.前述の通り,LQTSの中には安静時にQT間隔が正常範囲内の例がある.運動,情動ストレスや音刺激を契機とした一過性意識障害や痙攣の既往例,LQTSの家族歴のある例は,安静時の心電図だけで判断せず,積極的に運動負荷心電図やHolter心電図でQT延長を評価すべきである.
6. リスク評価
性別,年齢,QTc,初発年齢によりリスクが異なる.性別,年齢による違いの一因に性ホルモンの関与がある.テストステロンはQT間隔を短縮し,不整脈を抑制,エストラジオールはQT延長させ不整脈を誘発する12).
心イベントリスクは胎児期・乳児期では性差はないが,1歳以降思春期前では男児が高く,女児の5倍である13).思春期以降20歳までは性差はなく14),18~40歳では女性が男性の3倍になる15).
遺伝子型別で心イベントリスクを検討した我々の報告では,LQT1では1~12歳で男児のリスクが高く,LQT2では13歳以上で女性のリスクが高かった.特に,LQT2の女性は思春期になる前後で心イベントのリスクが高かった16).また胎児期・新生児期でも遺伝子型でリスクが異なる.LQT1は胎児期には徐脈が多く,新生児期の状態は良いことが多い.一方LQT3では胎児期にはTdPを含む胎児頻拍や房室ブロックを認めることがあり,そのような例は出生後の経過も悪い17).
QTcが延長しているほどリスクが高い.国際的な研究での20歳未満例のリスクの検討では18),QTc≧500が初回失神の予測因子であった.一方,遺伝子異常を認めるがQTcが正常範囲(<440 ms)の心イベントリスクは0.13%/年と低く,10歳未満で心イベントを起こした例はなかった19).
早期心イベント発生も危険因子である.7歳以下で心イベントが発生し,β遮断薬による治療でも失神を反復する例は高リスクで,1歳未満で失神を起こす例は特に重症である6).
また遺伝子変異部位によってもリスクは異なる.LQT1では膜貫通領域やCループ領域の変異が高リスクで,LQT2では膜貫通領域のミスセンス変異が特に高リスクである6).
7. 治療
日循のガイドラインでは小児期の薬物療法,非薬物療法も成人に準じている6).患者ごとにリスク評価を行い,治療方針を決定していく.
1)生活管理
LQT1やLQT2では交感神経活動刺激が誘因となり心イベントが起こりやすい.特にLQT1は運動,なかでも水泳中の心イベントが特徴的であり,マラソンでの心イベントも多い.具体的な管理は,学校心臓検診のガイドライン11)や学校心臓検診の実際20)を参照いただきたい.さらに自動体外式除細動器の使用を含めた事後対応も重要である.またQT延長作用のある薬剤を控えることも大切である.小児で特に問題になるのは注意欠陥多動性障害の治療薬やマクロライド系抗菌薬である.
2)薬物療法
β遮断薬内服は,心イベントのリスクは有意に低下させる.1~12歳のLQTS 3015例を対象とした報告でも,β遮断薬が心イベントリスクを53%抑制していた13).特にLQT1とLQT2では有効性が確立され,LQT3も女性での有効性が報告されている.β遮断薬の中でもβ1非選択性がβ1選択性よりも有効性が高いとされている21).LQT3には遅延性INa遮断薬のメキシレチン投与が有用性とされている22).LQT1-3以外の薬物療法の報告は限られているが,LQT7にはフレカイニドが23),またLQT8にはCa拮抗薬24)が有効との報告がある.
3)非薬物療法
植え込み型除細動器(ICD)植え込み適応に小児独自の基準はなく,成人の基準を参考とするが,体格の問題,不適切作動や合併症の多さなどから,成人の基準をそのまま当てはめるのには問題ある.特にLQT3の1次予防のためのICD植え込みは慎重に行うべきである.
ペースメーカは,徐脈が増悪因子となりTdPをきたす例が適応となる,またβ遮断薬服薬のため徐脈による症状が出現した場合もペースメーカは有効である.
左交感神経節切除術は日本では保険適応外であり,ほとんど行われていないが有用性は期待される.
1. 概要
QT短縮症候群(short QT syndrome: SQTS)は心電図でのQT間隔の短縮が特徴の遺伝性不整脈で,2000年に初めて報告された.VT, VFや心房細動(AF)が惹起され,失神や若年突然死の原因となる.心筋活動電位に関連したイオンチャネル遺伝子異常が原因として知られているが,遺伝子異常が同定されない症例が半数以上ある25).
2. 疫学
現在までに約100例の症例報告がある.男性が多く,男女比は7 : 3であった25).心停止例は約4分の1で,うち30歳以下が8割以上を占めた25).日本の小児について調べえた範囲では,遺伝子異常や症候を伴い,15歳未満で診断されたのは8例であり25),非常に限られている.発症年齢に性差があり,男性では10~20歳代での発症がほどんどだが,女性では発症年齢の偏りはなかった26).このような男女差は性ホルモンの影響が示唆される.報告が限られており,小児特有の臨床像で明らかなものはない.
3. 病態生理
SQTSでは遺伝子異常により心筋活動電位を形成するCaチャネルの機能低下やKチャネルの機能亢進が起こり,再分極が早まる.心筋細胞レベルでは活動電位持続時間が短縮し,心電図上ではQT間隔が短縮する(Fig. 1c).不均一な再分極に伴うphase 2 reentryがVFやVTを引き起こすと考えられ,LQTSに似る5).また有効不応期が短いため心房で複数のリエントリーが成立しやすく,AFを起こしやすい.
4. 遺伝的背景
現在までに6の原因遺伝子が明らかになっている(Table 4).遺伝子型と表現型の関連も一部にある.SQT2のKCNQ1-V141M変異例は胎児新生児期の徐脈と若年発症AFが主体で,QT短縮のない洞機能不全例や心電図異常のない例もあった.他の遺伝子異常では徐脈例は稀で,浸透率も高く,臨床像が異なった25).
Table 4 Genes causing short QT syndromes| Genetic Subtype | Disease-associated gene | Effect on channel function |
|---|
| SQT1 | KCNH2 | ↑IKr |
| SQT2 | KCNQ1 | ↑IKs |
| SQT3 | KCNJ2 | ↑IK1 |
| SQT4 | CACNA1C | ↓ICa-L |
| SQT5 | CACNB2b | ↓ICa-L |
| SQT6 | CACNA2D1 | ↓ICa-L |
5. 診断
1)症状
心イベントは,安静時や就寝時の発症が約8割である27).初回心イベントが突然死の症例もあり,乳児突然死症候群の原因としても報告されている28).また約20%にAFは合併しており,動悸や易疲労を訴えることがある.AFはSQT1–4に報告があった25).約半数は無症状例であった.
2)診断基準
診断基準は示されているが,議論の余地を残している.2011年にGollobらが初めてSQTSの診断基準を提唱した29).QT延長症候群のSchwartzスコアのようにスコア化されている.QT間隔に加え,Jpoint-Tpeak間隔,既往歴,家族歴,遺伝子検査の所見を加味し,診断する.その後,診断基準は2013年にHRS・EHRA・APHRS合同ステートメントで発表され(Table 5)7),2015年に欧州心臓病学会のガイドラインで示された30).この2つではQT短縮所見のみでもSQTSと診断しえるため過剰診断する可能性がある.HRS・EHRA・APHRS合同ステートメントではQTc≦330 ms,欧州心臓病学会ガイドラインではQTc≦340 msがSQTSとなる.この点以外は2つの診断基準はほぼ同じである.いずれの診断基準でもBazettの心拍補正を用いている.日循ガイドラインでは,SQTSの診断基準は示されていないが,男性ではQTc<330 ms,女性ではQTc<340 msの場合はSQTSを疑う必要があると述べている6).
Table 5 Expert consensus recommendations on SQTS diagnosis7)| 1. SQTS is diagnosed; |
| QTc (Bazett) ≦330 ms |
| 2. SQTS can be diagnosed; |
| QTc (Bazett) <360 ms |
| and one or more of the following: |
| ①a pathogenic mutation |
| ②family history of SQTS |
| ③family history of sudden death at age ≦40 |
| ④survival of a VT/VF episode in the absence of heart disease |
| Red font indicates differences from the European Society of Cardiology guideline30). |
3)心電図診断
QTcによるQT短縮の評価が最も重要であるが,以下の注意点がある.QTcは心拍補正法により異なる.Bazett補正では徐脈時はQT間隔を過剰補正してSQTSを過剰診断,頻脈時にはQT間隔を過小補正してSQTSを過小診断する懸念がある.一方Fridericia補正では頻脈時に過剰診断する懸念がある.また頻脈時には正常者とQT間隔がオーバーラップする可能性ある31).このために心拍数60~80/分での評価が推奨されている.
またT波形も重要な所見で,T波形は遺伝子型で異なることが知られている.SQT1では高く,左右対称性なT波形が多い.SQT2は様々であるが,SQT3では左右非対称なT波の報告が多い.詳細は別稿を参照いただきたい25).
4)負荷試験・ホルター心電図
運動負荷試験やホルター心電図においてはQT間隔が短縮し,正常に比し心拍数の変化に伴うQT間隔の変化が乏しいのが特徴である31).ホルター心電図はAFの評価にも有用である.
5)学校心臓検診での注意点
学校心臓検診のガイドラインでは,Bazett補正でQTc<330 msを1次検診の抽出基準としているが,小児でQT短縮と診断する基準は未確立であるとコメントがつけられている.健常な小児,特に思春期の男性ではQT短縮の頻度が高い.日本の学校心臓検診の報告では,男性の中学校1年生,高校1年生のQTc(Bazett補正)の1パーセンタイルは,それぞれ337 ms, 325 msであり32),成人中心のデータをもとに作成された診断基準では過剰診断となる可能性が高い.我々の経験では,学校心臓検診で抽出されたQT短縮例に繰り返し心電図を施行すると常にQT短縮を認める例は少なかった.前述の心電図診断の項で述べたQT間隔の過剰補正がその一因と推察された(未発表データ).
6. リスク評価
リスク評価法は確立されていない.QT短縮の程度と重症度の関連は示されておらず,特に無症候例ではQT短縮の程度によるリスクの層別化には否定的な報告が多い.
7. 治療
生活管理や治療に関するエビデンスは乏しく,運動制限の有効性は示されていない.
日循のガイドラインではキニジンを第一選択薬に上げている6).特にSQT1でQT間隔が正常化する例があり心イベント抑制効果が期待される26, 33).ソタロールなどのIII群薬はSQT1に対するQT延長効果は乏しいが,他の亜型で効果が期待されている.
ICDが突然死防止に有用であるが,QTが極端に短縮しているため,T波をQRSと認識し誤作動する場合がある.日循のガイドラインでは6),心肺蘇生の既往例と自然発作の持続性VT/VF既往例にICD植え込みを推奨し,突然死の家族歴のある無症状例には有用性が確立していないとしている.
1. 概要
カテコラミン誘発多形性心室頻拍(catecholaminergic polymorphic ventricular tachycardia: CPVT)は1960年に初めて報告された遺伝性不整脈である34).若年者に多く,運動や情動ストレスが契機となって失神や突然死を引き起こす.安静時心電図での異常が少なく,交感神経刺激に伴い心室不整脈が出現する.心筋細胞内のCa過剰に伴う遅延後脱分極(DAD)が不整脈の発生に関与している.現在まで複数の遺伝子異常が報告されている.
2. 疫学
CPVTの推定頻度は10,000人に1と言われている6).我が国の集計では家族発症が6%であった.また性差はないとされているが,我が国の集計では女性が約3分の2を占めた35).若年で致死的な心イベントを起こすことが多い.初発症状が出現する年齢は7~12歳であり,40歳までに30%のCPVT患者が心停止や突然死を起こす.
3. 病態生理
心筋細胞内Ca2+動態に関連する蛋白の遺伝子異常が原因で細胞内Ca2+増加が過剰となる.するとNa/Ca交換系のforward mode(1個のCa2+が細胞外へ,3個のNa+が細胞内へ移動)が活性化され,一過性内向き電流によるDADを機序とする不整脈が起こる.交感神経刺激時にはリアノジン受容体やL型Caチャネルが活性化し,細胞内Ca2+がより増加するため不整脈が起きやすくなる.
4. 遺伝子的背景
遺伝子解析が進み亜型に分類され,遺伝子型と表現型の関連が明らかになってきている.主な遺伝子型について下記の通り述べる.
CPVT1はリアノジン受容体遺伝子RYR2の変異が原因である36).リアノジン受容体は筋小胞体に存在するCa2+放出チャネルで,筋小胞体内に貯蔵されたCa2+は収縮期にリアノジン受容体から細胞質に内に放出される.CPVT1ではリアノジン受容体の機能亢進により拡張期にもCa2+が細胞質内に漏出する.そのため細胞内Ca2+濃度が上昇し,DADが起こる.CPVTの約半数をしめ,常染色体優性遺伝をするが,日本の報告では約57%は孤発例であった37).約10%が突然死する.
CPVT2はカルセクエストリン2遺伝子CASQ2の変異が原因である38).カルセクエストリンは筋小胞体内に存在するCa結合タンパクで,リアノジン受容体を安定化する.またCa貯蔵の役割もある.CPVT2ではリアノジン受容体安定化の阻害による筋小胞体からのCa漏出やCa貯蔵の障害により,DADが起こる.CPVTの約1%を占める.10歳までに全例発症し,突然死が約40%と高率である.
さらにCPVT関連疾患として以下の2つがある.LQT4はアンキリンBをコードする遺伝子ANK2の異常が原因である.QT延長を認め,AFや徐脈,房室ブロックを起こす39).アンキリンBは細胞膜構造タンパクでイオンチャネルを細胞膜につなぎとめる役割があり,Na/Ca交換系などに様々な影響を与える.QT延長をきたさず,運動負荷でCPVTを呈した例,洞機能不全を呈した例なども報告され,「アンキリン症候群」と呼ぶことある40).LQT7の原因遺伝子であるKCNJ2の変異でも,QT延長を認めず,CPVTを呈した症例が報告されている41).
5. 診断
1)症状
運動中の失神や突然死が典型的であるが,情動ストレスや入浴が関連することもある.乳児突然死症候群の原因にもなりうる.安静時の心電図が正常なためてんかんとして管理され,診断が遅れることもある.
2)診断基準
日循のガイドラインもHRS・EHRA・APHRS合同ステートメントの診断基準に従っている(Table 6)6, 7).交感神経刺激で誘発される特徴的な心室不整脈や遺伝子変異が診断の鍵となる.
Table 6 Expert consensus recommendations on CPVT diagnosis7)| 1. CPVT is diagnosed in the presence of a structurally normal heart, normal ECG, and unexplained exercise or catecholamine-induced bidirectional VT or polymorphic ventricular premature beats or VT in an individual <40 years of age. |
| 2. CPVT is diagnosed in patients (index case or family member) who have a pathogenic mutation. |
| 3. CPVT is diagnosed in family members of a CPVT index case with a normal heart who manifest exercise-induced premature ventricular contractions (PVCs) or bidirectional/polymorphic VT. |
| 4. CPVT can be diagnosed in the presence of a structurally normal heart and coronary arteries, normal ECG, and unexplained exercise or catecholamine-induced bidirectional VT or polymorphic ventricular premature beats or VT in an individual >40 years of age. |
3)心電図診断
安静時心電図では,徐脈傾向,U波が目立つなどのわずかな異常を認めることがあるのみで,CPVTの診断は困難である.心室不整脈は交感神経緊張時に出現する.QRS軸が180度変化する2方向性VTが特徴的で,多形性となることもある.しかし誘導によっては2方向性VTが確認できない場合や,軽症例では心室期外収縮の単発や2段脈のみ認める場合もある.運動時や興奮時に失神,心停止した若年者では先天性LQTSとの鑑別が困難な症例があるが,我々の検討から,蘇生直後のQT間隔は診断価値が乏しく,運動負荷終了直後にQT短縮する例はCPVTの可能性が高いことが示唆された42).
4)ホルター心電図・負荷試験
運動負荷試験が診断に最も有用である.運動強度の増加と心拍数の上昇に伴い,上室頻拍や心室頻拍が出現し,頻度が増すのが典型的である.さらにVFとなる場合もある.ホルター心電図は,運動負荷ができない乳幼児や情動ストレス時の評価に有用である.エピネフリン負荷試験も運動負荷ができない症例には有用かもしれない.
5)遺伝子検査
日循のガイドラインでは,不確定な症例には重要な診断方法であり,強く推奨しているが6),まだ保険適応はない.
6)学校心臓検診での注意点
安静時の心電図での異常は少ないため,心電図での抽出が困難な疾患であるが,家族歴,既往歴に注意する.若年突然死の家族歴や,運動や情動ストレスを契機とした一過性意識障害や痙攣の既往がある例には,安静時の心電図だけで判断せず,積極的に運動負荷心電図やHolter心電図を行うべきである.
6. リスク評価
エビデンスはまだ乏しいが,心停止の既往が最もリスクが高い43).さらに低年齢での診断例もリスクが高い.
7. 治療
日循のガイドラインもHRS・EHRA・APHRS合同ステートメントに準じている6).
1)生活管理
運動制限はCPVT全例に必須で,さらにストレスを避ける配慮も必要である.
2)薬物療法
β遮断薬が第一選択薬で,次にフレカイニドが推奨される.β遮断薬は有症状のみならず,遺伝子異常を持つ無症状者も含めCPVT全例に第一選択薬として推奨される.β遮断薬の中でもナドロールなどの長時間作用薬が推奨される.CPVT101例を経過観察した報告では43),β遮断薬を内服していないことが,心イベントの独立した予測因子であり,早期の治療が予後を改善させる可能性が示唆される.フレカイニドはβ遮断薬内服でも失神やVTを繰り返す症例に勧められる.ベラパミルをβ遮断薬に追加する効果は限定的である.
3)非薬物療法
ICDは,可能な内科治療をすべて試みても効果がない例に推奨される7).ただし,不適切作動に伴う痛みでVTが誘発される可能性がある.特に小児に対するICD植え込みは長期予後が不明であり,慎重にすべきである.近年のシステマティックレビューでは44),CPVTの約3割にICDが植え込まれ,その約半数が1次予防目的であった.ICD植え込み症例の40%に適切作動があったが,20%に不適切作動があり,約1%はICD作動が誘因となったエレクトリカルストームで死亡していた.
交感神経切除術は,β遮断薬内服下でも失神,VTを繰り返す症例やβ遮断薬内服が困難な例は考慮してもよいかもしれないが7),日本での保険適応はない.
1. 概要
Brugada症候群(BrS)は, 1992年に初めて報告されたVFをきたす遺伝性不整脈である45).12誘導心電図の右胸部誘導に特徴的なST上昇を認める.この所見はBrugada心電図とよばれるが,時に正常化する.若年から中年男性が夜間にVFを引き起こして突然死するのが典型例で,我が国のぽっくり病の原因と考えられている.SCN5Aなどの遺伝子異常の報告があるが,Brugada心電図やVF発生の機序には議論の余地がある.
2. 疫学
人種差があり,東南アジアに多い.成人を中心としたまとめでは男性が多く,本邦の報告では男性が95%を占める.Brugada心電図が初めて出現するのは30~40歳台が多く,突然死発生の平均年齢は50歳台である.一方,小児期には少なく,学童でのBrugada心電図の頻度は0.005%程度と報告されている46).また小児期では頻度の性差はほとんどない47).小児症候例は少ないが,乳児期に発症する症例もあり48),症候例の予後は悪い47).
3. 病態生理
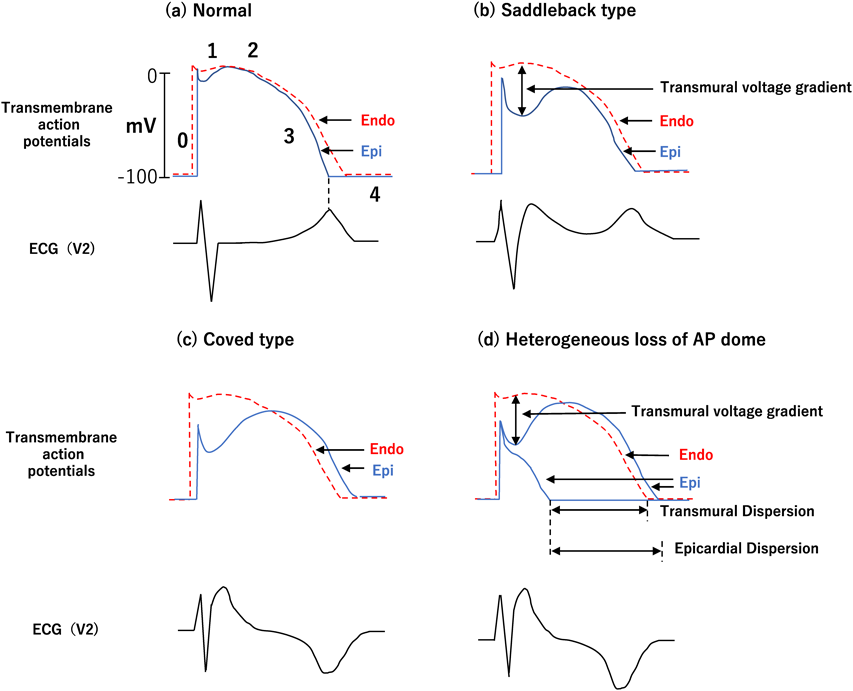
BrSの本体が,右室流出路心筋壁における再分極の不均一性であることが示唆され49),Brugada心電図の特徴であるコブド型やサドルバック型のST上昇や催不整脈性の有力な成因論となっている.
右室流出路の心内膜(endo)と心外膜(epi)の活動電位波形には違いがあり,その電位差を12誘導心電図の右胸部誘導が記録していると捉えることができる.endoがepiより電位が高いと心電図の電位は陽性(上昇)に,低いと陰性(低下)となる.endoが先に脱分極するが,先に再分極するのはepiのため,活動電位持続時間はepiが短い.またepiがendoに比しItoが発達している.この違いは特に右室流出路で大きく,そのためendに比べepiで第1相から第2相でのノッチとドーム(AP dome)形成が目立つ.しかしその電位差は比較的わずかなためSTはほぼ基線に記録される(Fig. 5a). BrSでは相対的な内向き電流の減少(内向き電流の減少や外向き電流の増加)で,epiでのノッチが大きくなり,endoとの電位差が開きサドルバック型のST上昇が起こる(Fig. 5b).相対的な内向き電流のさらなる減少でL型カルシウムチャネルの活性化が遅れ,ドーム形成が遅延し,endよりもepiの再分極が遅れるためT波が陰転化し,コブド型のST上昇に変化する(Fig. 5c).またさらに相対的な内向き電流が減少すると,L型カルシウムチャネルが活性化されず,epiの第2相のドームが消失し,epiが早期に再分極する(Fig. 5d).するとepiおいて電気的なばらつき(epicardial dispersion of repolarization)が増大するためphase2 reentryが起きる.ICaLは自律神経の影響を受け,交感神経緊張低下や迷走神経緊張亢進で減少し,ST上昇とVFが誘発されると考えられている.
一方,右室流出路付近での伝導遅延などの脱分極異常が主な成因であるという意見もある50).
4. 遺伝学的背景
SCN5AにNaチャネルの機能低下をもたらす遺伝子異常が報告された(BrS1)51).Naチャネルの機能低下が心室筋の脱分極を障害し,再分極では相対的な内向き電流の減少を起こして病態に関与すると推察され,イオンチャネル病として捉えられた.現在では23の遺伝子異常(BrS1–23)が明らかになっているが,BrS1が15~30%と最も頻度が高い.
しかし近年では,イオンチャネル病として捉えるだけでは説明がつかず,複数の修飾遺伝子や,炎症,繊維化など後天的要因・環境要因を含めて捉えるようになっている.
遺伝子型と表現型の関連については,近年本邦からSCN5A変異と予後の関係が報告された52).この研究では,SCN5A変異陽性群が陰性群に比し,初回心イベントの発生年齢が有意に低く,心イベント発生率が高かった.
5. 診断
1)症状
失神,めまい,苦悶様呼吸,動悸,胸部不快感などがあり,安静時,睡眠時,食後に多い.特に小児では,発熱時に発症することが多く53),熱性痙攣と誤診される例がある.
2)診断基準
日本循環器学会のガイドライン6)ではBrSを心電図所見で診断している.A:自然発生(正常肋間あるいは高位肋間記録)でのタイプ1 Brugada心電図,B:発熱による誘発(正常肋間あるいは高位肋間記録)でのタイプ1 Brugada心電図,またはC:薬剤負荷でタイプ1に移行したタイプ2またはタイプ3 Brugada心電図をBrSと診断する.さらに臨床歴の有無で有症候性BrSか無症候性BrSに分ける.
3)心電図診断
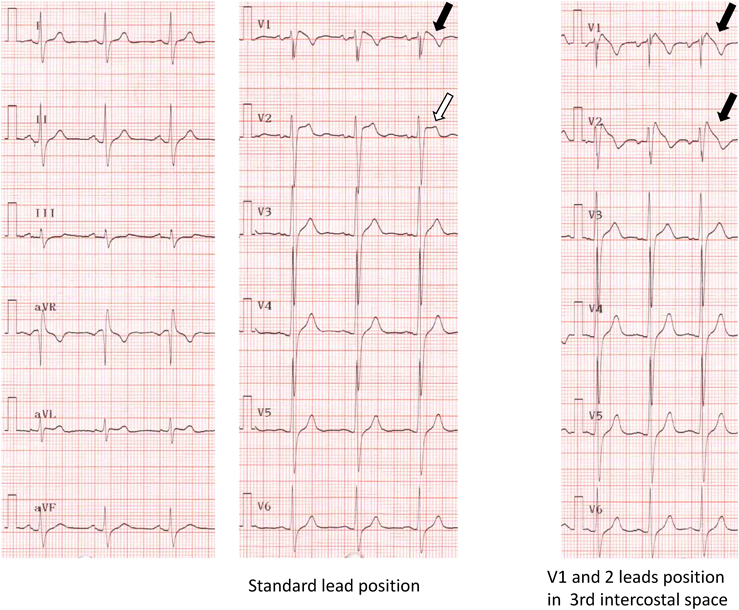
Brugada心電図のST上昇はコブド型とサドルバック型に分類される.コブド型は,J点付近のST上昇を頂点として,水平部分をほとんど認めずに陰性T波に移行し(Fig. 6),サドルバック型は,J点付近で上昇したSTが徐々に低下するが,基線まで復さずに引き続き陽性または2相性T波に移行する(Fig. 6).
またBrugada心電図はタイプ1~3に分類される54, 55).タイプ1はJ点で2 mm以上のST上昇のあるコブド型,タイプ2はJ点で2 mm以上のST上昇のあるサドルバック型で,かつJ点から引き続くST部分が1 mm以上を保ったままT波に移行するもの,タイプ3は,J点のST上昇が1 mm未満でサドルバック型またはコブド型のものである.J点で1~2mmST上昇を示すものは分類されていない.
通常の右胸部誘導(V1, 2)の電極位置ではBrugada心電図を示さないが,第2肋間までの高位肋間記録で顕在化することがあるのでBrSを疑う場合は必ず,V1, 2の高位肋間記録をとる(Fig. 6).前述通り診断基準にも採用されている.
4)学校心臓検診での注意点
学校心臓検診のガイドラインでは11),タイプ1 Brugada心電図を抽出すべき所見とし,J点が0.2 mV以上のサドルバック型Brugada心電図を単独では必ずしも抽出しなくてもよい所見としている.Brugada心電図は正常化することがあり,1回の心電図でBrSを鑑別することはできない.安静時や睡眠中の若年突然死やBrSの家族歴,熱性けいれんや安静時の一過性意識障害の既往歴,洞機能不全,心房不整脈や伝導障害などの所見を持つ例には注意し,胸部誘導の肋間上げ心電図を積極的にとるべきである.
5)ホルター心電図・負荷試験
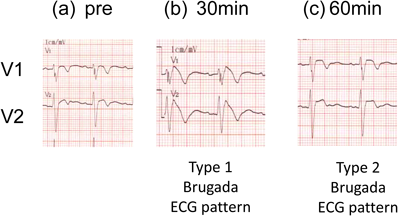
BrSにおけるST上昇には日差や日内変動があり,正常化している時もある.夕食後や夜間にBrugada心電図が顕在化することが多いといわれ,12誘導ホルター心電図で評価できる56).また負荷試験もBrugada心電図の顕在化に有用である.負荷試験には薬物負荷,運動負荷,経口糖負荷や満腹試験がある.運動負荷試験や満腹試験など,より生理的な方法でタイプ1 Brugada心電図を確認すること優先させ,それでも診断がつかない場合に薬剤負荷試験を行うべきである.薬剤負荷にはIaおよびIc群のNaチャネル遮断薬(ピルジカイニドなど)が用いられ,負荷後にタイプ1 Brugada心電図を認めると陽性と判断される.
運動負荷では交感神経刺激によりBrugada心電図は改善するが,負荷終了後に副交感神経優位となり,Brugada心電図が顕著になることがある57).経口糖負荷試験58)や,短時間に多くの食物を摂取する満腹試験59)でもBrugada心電図が顕在化することがある(Fig. 7).
6. リスク評価
成人例を中心とした検討がなされ,臨床所見の重要性が示唆されている.心肺停止やVFの既往例が最もリスクが高く,不整脈源性失神の既往例のリスクは心停止やVFの既往例の1/4,無症候例のリスクは不整脈源性失神の既往例の1/4とされている60, 61).無症候例の年間イベント発生率は0.5%と低いが61, 62),イベントの多くは心停止または突然死である.さらに無症候例では,臨床歴,家族歴,遺伝子検査結果をリスク評価の参考となる6).小児では,欧州のBrS 128例の検討した報告があり47),症候例に洞機能不全,心房不整脈や伝導障害の合併が多かった.また予後が悪いのは,症候例と自然発生のタイプ1心電図を示す例であった.我が国のBrS小児例の予後やリスクは不明であり,その解明が待たれる.
7. 治療
1)生活管理
小児では発熱が心イベントの誘因となることが多いため発熱時は解熱剤を積極的に使用し,解熱をはかる.
2)薬物療法
薬物療法はICDに比較して不確実であるが,ICD植え込み適応があるができない例や,ICD植え込み例のVF予防のために必要である.VF予防として最もエビデンスがあるのはキニジンで,VFストームではイソプロテレノールが有効である.
3)非薬物療法
ICD植え込みがBrS患者の突然死予防の有効性を証明された唯一の治療法である.日循のガイドラインでは6),心肺停止蘇生歴またはVF既往のタイプ1 Brugada心電図例はICD植え込みのクラスIの適応であり,タイプ1 Brugada心電図に加えて不整脈原性失神(尿失禁を伴うことが多く,非不整脈原性失神に特徴的な誘因を伴わない)あるいは苦悶様呼吸を有する例はクラスIIaのICD適応とされた.成人例を中心とした研究でICD植え込んだBrSの適切作動は年間2.6%であったのに対し,不適切作動を含むICD関連合併症の発生率が8.9%と高いと報告された63).小児ではさらにICDの合併症が多い,成人のデータに基づくICD植え込み適応を当てはめるのは慎重になるべきである.
カテーテルアブレーションの有効性も報告され始めたが,日循のガイドラインでは薬物抵抗性VFストーム症例やICD頻回適切作動例への有用性が確立していないとしている.小児例は限られているが,ICD植え込みと同時にアブレーションを行った3歳例が2018年に報告された64).