染色体22q11欠失症:発見と合併心疾患Chromosome 22q11 Deletion Syndrome: Discovery and Associated Cardiovascular Anomalies
東京女子医科大学循環器小児科Department of Pediatric Cardiology, Tokyo Women’s Medical University ◇ Tokyo, Japan
発行日:2017年1月1日Published: January 1, 2017
高尾教授は1976年にFallot四徴症と特異顔貌と鼻声の合併として円錐動脈幹異常顔貌症候群を発見し,1984年にはこの症候群に胸腺低形成と免疫不全があることを発見した.1992年にはBurn教授との共同研究でこの症候群の染色体22q11の微細欠失を証明した.1993年には末梢血リンパ球FISH法で22q11欠失検査を開始し,22q11欠失症の先天性心疾患の全体像が明らかになった.本症候群の80%に先天性心疾患が合併し,その内訳は多い順に各種のFallot四徴症(約30%),大動脈弓離断(約15%),総動脈幹残遺(約15%),心室中隔欠損症(約15%)である.本症候群の先天性心疾患には特徴的に大血管異常が合併する.即ち,Fallot四徴症の半数は肺動脈閉鎖を合併し,その大部分で動脈管を欠如して体肺側副動脈(MAPCA)を合併する.本症のFallot四徴症には大動脈とその分枝動脈の異常(右側大動脈弓,鎖骨の高さに達する大動脈弓,動脈管欠損,肺動脈弁欠損,MAPCA, 鎖骨下動脈起始異常)が合併する.本症の大動脈弓離断はA型でなく,全てB型である.本症の総動脈幹残異では肺動脈低形成,肺動脈の交差性起始,MAPCAをしばしば合併する.本症の心室中隔欠損に大動脈異常,血管輪が時に合併する.
The late Dr. Takao (1925–2006), professor emeritus of Tokyo Women’s Medical University, discovered the conotruncal anomaly face syndrome in 1976. This syndrome is a combination of dysmorphic facial appearance, nasal speech, and congenital conotruncal heart anomalies. Dr. Takao later discovered the association of thymic hypoplasia and immune deficiency with this syndrome. In 1992, he and Dr. Burn identified a chromosome 22q11 deletion (del22q11) in five Japanese patients with this syndrome. Since 1993, data from 200 patients with del22q11 have been collected, and detailed cardiovascular studies have been performed at his hospital, which have helped to identify the associated cardiovascular anomalies that typically present with this syndrome. Eighty percent of patients with del22q11 syndrome have congenital heart diseases, including tetralogy of Fallot (30%), interrupted aortic arch (15%), truncus arteriosus (15%), and ventricular septal defect (15%). In 50% of patients with tetralogy of Fallot, pulmonary atresia occurs, and the major aortopulmonary collateral artery (MAPCA) is typically present. Tetralogy of Fallot cases with del22q11 is more commonly associated with arterial anomalies than tetralogy cases without the deletion. These associated aortic anomalies include a right aortic arch and an elongated high aortic arch. The associated arterial anomalies include absence of the ductus arteriosus, MAPCA, anomalous origin of the subclavian artery, isolation of the subclavian artery, isolation of a pulmonary artery (absence of a pulmonary artery), and aortic (Kommerell’s) diverticulum. Interruption of the aortic arch is type B in del22q11. Truncus arteriosus with del22q11 is associated with pulmonary artery hypoplasia, MAPCA, and crossing of the pulmonary arteries.
Key words: chromosome 22q11 deletion; conotruncal anomaly face syndrome; tetralogy of Fallot; truncus arteriosus
© 2017 特定非営利活動法人日本小児循環器学会© 2017 Japanese Society of Pediatric Cardiology and Cardiac Surgery
染色体22q11.2欠失症(以下,本症)はDiGeorge症候群,Velocardiofacial症候群(Shprintzen),円錐動脈幹異常顔貌症候群(高尾)を含む.私は2015年に名誉ある第1回本学会功労賞を受賞したが,本賞は御存命なら故高尾篤良名誉教授(1925~2006,東京女子医科大学心研小児科)(Fig. 1)がまず受賞されたはずである.本症はしばしば複雑なFallot四徴症や総動脈幹残遺を合併し,高尾教授定年(1990年)後の11年間に私の心研小児科の臨床で最も力を入れた研究テーマとなった.ここに高尾教授による円錐動脈幹異常顔貌症候群の発見と本症に合併する心疾患についてレビューする.

高尾教授の小児循環器学はHouston, Texas小児病院心臓病センターでの2年間の循環器小児科フェローで始まった1).この心臓病センターは1950年代当時Johns Hopkins病院(Blalock–Taussig手術(1944)の発祥の地)から来た外科のDenton Cooley教授,小児科のDan McNamara教授を擁し,世界の先天性心疾患手術の先端をいくセンターであった.高尾教授はMcNamara教授の厚い信任を得て,30年後にこの小児病院で小児心臓病学の大部の専門書を編集出版する際には心室中隔欠損症の章の執筆を依頼されることになる1).帰国後1958年に東京女子医科大学日本心臓血圧研究所(心研)外科の助手になった2).次いで心研小児科を1人で立ち上げ,数年後には教授になった2).当時心研には全国から先天性心疾患の患者が集まり,とくに当時手術成績の不安定であったFallot四徴症の小児が殺到していた.高尾教授の週2回の外来は朝から夕方遅くまで昼食をとる時間もないほどこれらの患者で混み合っていた.高尾教授は診察室ではよくこどもに話しかけていた.1970年代はじめにはFallot四徴症のこどもの中に特有の顔貌と鼻声をもつ1群があることに気がついた.総動脈幹残遺の例にもこの顔貌があり,その他の先天性心疾患にはないので,この症候群を円錐動脈幹異常顔貌症候群conotruncal anomaly face syndromeと命名した3).この発見は1976年に雑誌小児科の巻頭の『目で見る小児科』欄に顔写真として発表された3).高尾教授は本症候群の本態解明のため,親しいBostonのVan Praagh教授(高尾教授の指導による1970年から2年の安藤正彦教授の留学先)と相談し,産経新聞社明美ちゃん基金の後援を得て,1978年に先天性心疾患の形成と成因の国際シンポジウムを開いた.このシンポジウムの記録(1980)4)が高尾症候群発見の英文論文であり,50症例が報告されているが,46例がファロー四徴症とその極型,2例が総動脈幹残遺,2例が両大血管右室起始症であった.残念なことにシンポジウムのproceedingの常で,この論文は国際的に広く読まれるには至らず,高尾症候群の名は日本国内と高尾シンポジウムの参加者の間までにとどまった.
この報告より13年前,1965年にPhiladelphia, Pennsylvania大学の小児内分泌学者DiGeorge教授が先天性の胸腺欠如と副甲状腺欠如と低カルシウム血症,免疫欠損の合併例を報告5)し,この組み合わせはDiGeorge症候群と呼ばれていた.1978年にはNew Yorkの言語音声学者Shprintzenが粘膜下口蓋裂,鼻声,先天性心疾患,特異顔貌の組み合わせをVelocardiofacial syndromeとして発表した6).門間は1970年に高尾教授に誘われて東大小児科助手から心研小児科講師になった.当時心研では小児科と外科は同じ医局で,門間も高尾教授も希望して手洗いして第2助手として手術に参加することがあった.第1回高尾シンポジウム開催のころには高尾教授は本症候群のFallot四徴症の小児の手術で開胸すると胸腺が小さく,全く欠如することもあることに気がついた.更に北大小児科から研修に来た清水隆君に調べさせてこれらの小児で胸腺由来の免疫能の低下が確認され,第2回高尾シンポジウム(1983)で発表された7).DiGeorge症候群でも先天性心疾患と特異顔貌を合併することがわかってきて,高尾症候群とDiGeorge症候群の類似性が明らかになった.DiGeorge症候群の成因(染色体異常)の研究が1980年代に進み,染色体22q11.2の微細欠失があることが染色体分染法で報告され8)(1981),Pennsylvania大学遺伝学Emanuel教授も1982年にこれを確認9)して22番染色体のDiGeorge locusが確定した.
1990年台初頭には遺伝子プローブ,FISH法を用いる染色体分析が始まり,DiGeorge症候群とShprintzen症候群で80~90%に染色体22q11の微細欠失が存在することが1992年に報告された10, 11).高尾教授もこうした新しい方法で染色体分析を行うべく,BostonでVan Praagh教授とNadal-Ginal教授のもとに留学してきた松岡瑠美子講師(後,特任教授)に指示して心研地下に国際分子細胞免疫施設をつくり,1990年高尾教授定年と同時にこの施設が稼働しはじめ,高尾教授も定年後は毎週施設長としてここに来られてカンファレンスを主導されていた.しかしここでもはじめは,高尾症候群のDiGeorge locus検索には技術的にわずかに手が届かなかった.そのうちに1991年に高尾症候群と同じ顔貌を持つ症例を含む22q11欠失の報告12)が英国から出た.高尾シンポジウムで知り合いになっているNewcastleのBurn教授の名前もそこにあったので,高尾教授が早速手紙を書くと,たまたま東京で双生児研究の会があり,Burn教授も参加するとのことで,その機会に心研で高尾症候群の症例を一緒に診て,血液サンプルを持ち帰り,彼らのプローブで22q11欠失がないか確かめてもらった.その結果は全例に欠失があり,高尾症候群の染色体22q11.2の微細欠失が証明された.
この共同研究には次のような経緯で門間も加わった.高尾教授は寡黙で最小限必要なことしか言われない方であったが,この時も門間のところへ来て,細かい説明抜きで,John Burnが来るから何人でもいいですから患者を集めてください,と言われた.当時高尾症候群の診断名は心研小児科でも未だ使われていなかった.そこで私は当時の私の臨床研究テーマであった肺動脈閉鎖とMajor Aorto-Pulmonary Collateral Artery(MAPCA)を持つFallot四徴症のリストからそれらしい顔を持つ5名を選び,自分で患者の親に電話をして指示された日の午後に心研外来に来てもらった.ありがたいことに5人全員が来てくれて,Burn,高尾,門間で診察し,顔写真をとり,10 mLずつ採血し,Burn教授は翌日の飛行機で血液をLondonに持ち帰り,仲間の研究者が染色体probe D0832でDiGeorge locusを調べ,5名全員にその欠失を証明した.Burn教授はこの結果を論文にして初めLancetに投稿したが,Lancetには既にDiGeorge症候群とShprintzen症候群の22欠失の論文10)が出ており,返されたので,Journal of Medical Geneticsに送り,1993年に同誌に掲載13)された.これで高尾症候群の染色体微細欠失が確定した.Burn教授からの欠失検査結果の報告を高尾教授を通じて私が受け取ったのは採血の4か月後で,その4か月間私は大学入学試験後に合格発表を待った20日間と同じく,期待と不安でひどく落ち着かなかった.
1993年には国際分子細胞免疫施設で我が国で最初に22q11欠失のFISH法による患者末梢血液検査が開始され,心研に多数蓄積されていた円錐動脈幹異常顔貌症候群の患者の血液検査が進み,1~2年のうちに22q11欠失の確定症例が100例を超え,女子医大小児科,耳鼻科,精神科などの協力も得て,本症候群臨床像の全容14–16)が明らかになった.私の外来は検査の手配で忙しかった.高尾教授は大学定年(1990年)の後は新宿駅に近い榊原記念クリニックで患者を診ておられたが,採血の患者負担を嫌って,ご自分の患者では血液検査を全くされなかった.
本症候群の22番染色体q11.2の微細欠失部には約30の遺伝子が含まれている.2001年2月にはこれら30の遺伝子のほぼ中央に位置するTBX1が心疾患を生じる責任遺伝子であることが確定17)した.心研の症例中にも少数ながら本症候群の諸症状を有しながらFISH検査(古谷喜幸博士担当)で欠失のない症例があり,これらの症例13例のTBX1を高尾–松岡の指導下に大学院生だった八木寿人博士が遺伝子解析をしたところ5例に点突然変異が見つかった18).ただしこの5例には知恵遅れは合併していなかった.
慶應義塾大学小児科山岸敬幸教授は高尾教授と親しく,国際分子細胞免疫研究施設にも出入りしていたが,高尾シンポジウムの縁でTexas大学Srivastava教授(現在UCSF, Gladstone Institute,岡山大学佐野俊二教授の2016年12月からの赴任先)のもとでTBX1の心臓発生初期の発現細胞を調べ,予想に反して神経堤細胞ではなく二次心臓形成領域の細胞に発現することを発見し19),本症候群の円錐動脈幹疾患は二次心臓形成領域細胞の異常によることが判明した.
FISH法で本症候群が確定診断できるようになり,その心臓血管異常の全容20)が明らかになった.Table 1に世界各地からの主な報告を示す.Parisの小児病院からの報告(Boudjemline)21)はユニークで,円錐動脈幹異常の胎児診断例で染色体22q11欠失の有無を調べ,欠失例54例を集計した.その結果はFallot四徴症が過半数を占め,大動脈弓離斷と総動脈幹残遺がこれに次ぐことを示した.あとの5報告は出生後の症例の検査結果であり,Ryanは欧州多施設の共同研究22),Parkは韓国での多施設共同研究23)を報告し,Matsuoka15), Marino24), Oskarsdottir25)は1施設から報告した.これらの報告では正常の心臓が約20%,Fallot四徴症は約25%を占める.肺動脈弁欠損を伴うFallot四徴症は重症で出生後の経過不良のため胎児診断例より少ない.同じ理由で総動脈幹残遺と大動脈弓離断例も胎児診断例より生存例では少ない.
| Cardiovascular anomaly | Boudjemline et al.21)* | Ryan et al.22)** | Matsuoka et al.15)** | Marino et al.24)** | Oskarsdottir et al.25)** | Park et al.23)** |
|---|---|---|---|---|---|---|
| N (%) | N (%) | N (%) | N (%) | N (%) | N (%) | |
| TN=54 | TN=545 | TN=183 | TN=88 | TN=100 | TN=222 | |
| Normal heart | 110 (20) | 23 (13) | 23 (26) | 36 (36) | 32 (14) | |
| Tetralogy of Fallot (TF) | 14 (26) | 95 (17) | 67 (39) | 23 (26) | 13 (13) | 59 (27) |
| TF, absent pulmonary valve | 6 (11) | 2 | 2 (1) | + | 3 (1) | |
| TF+PA+PDA | 11 (20) | 55 (10) | 4 (2) | 24 (27) | 5 (5) | 15 (7) |
| TF+PA+MAPCA | #1 | #1 | 45 (25) | #1 | #1 | 42 (19) |
| Ventricular septal defect | 75 (14) | 25 (13) | 15 (17) | 14 (14) | 39 (18) | |
| Interrupted aortic arch | 10 (19) | 74 (14) | 7 (4) | 8 (9) | 6 (6) | 10 (5) |
| Truncus arteriosus | 9 (17) | 51 (9) | 4 (2) | 10 (11) | 10 (10) | 1 |
| Pulmonary valve stenosis | 13 (2) | 1 (1) | ||||
| Atrial septal defect | 8 (1) | 2 (1) | 2 (2) | 4 (4) | 7 (3) | |
| Atrioventricular septal defect | 5 (1) | 1 | 1 | |||
| Double outlet right ventricle | 4 (1) | 3 (2) | 1 (1) | 7 (3) | ||
| Transposition of great arteries | 4 (7) | 4 (1) | 1 | 3 (3) | 1 (1) | |
| PDA | 7 (1) | 2 (1) | 3 (3) | 2 (1) | ||
| DCRV | 2 (2) | 3 (1) | ||||
| #1: included in TF+PA+PDA. +: included in TF. DCRV: double-chambered right ventricle. MAPCA: major aortopulmonary collateral artery. PA: pulmonary atresia. PDA: patent ductus arteriosus. TAPVC: total anomalous pulmonary venous connection. TF: tetralogy of Fallot. TN: total number. Data modified and cited with the permission of the author and the publisher from reference 20). | ||||||
臨床的には各種の先天性心疾患それぞれにおける染色体22q11欠失症の頻度が重要である.Fallot四徴症,大動脈弓離断,総動脈幹残遺,大血管転位における染色体22q11欠失症の頻度が報告20)されている.それによるとFallot四徴症の約20%,その肺動脈閉鎖,または肺動脈弁欠損の合併例では30%が欠失症である.大動脈弓離断では50%が欠失症である.総動脈幹残遺では30%が欠失症である.完全大血管転位では生後の検査では欠失例はほとんどないが,胎児例で12%と報告されていて,この違いの理由は不明である.
本症例では入院して造影検査などで心臓血管異常を詳しく調べると,非欠失例には少ない血管異常(Figs. 2, 3, 4)をしばしば合併することが明らかになった26–30).
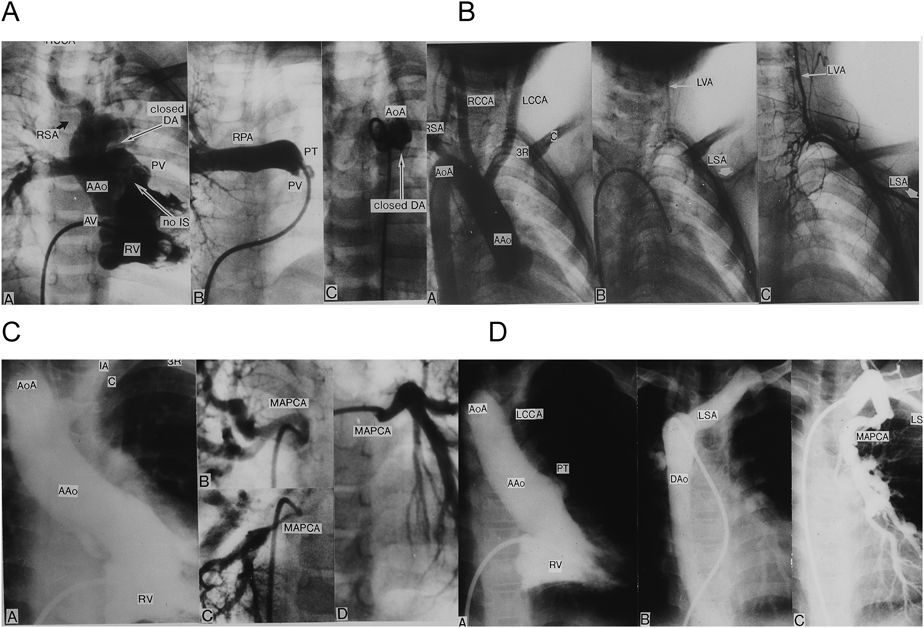
(A) TF, conus septal defect, aberrant right subclavian artery (RSA), and isolation of the left pulmonary artery (absent left pulmonary artery). (B) TF, pulmonary atresia (PA), right aortic arch (RAA), high aortic arch (HAA), and isolation of the left subclavian artery. (C) TF, PA, RAA, HAA, and the major aortopulmonary collateral artery (MAPCA). (D) TF, PA, MAPCA, and RAA. Aberrant left subclavian artery (LSA). AAo: ascending aorta, AoA: aortic arch, AV: aortic valve, DA: ductus arteriosus, DAo: descending aorta, F: frontal, IA: innominate artery, IS: infundibular septum, L: lateral, LCA: left coronary artery, LCCA: left common carotid artery, LPA: left pulmonary artery, LSA: left subclavian artery, LVA: left vertebral artery, MPA: main pulmonary artery, PT: pulmonary trunk, PV: pulmonary valve, RCCA: right common carotid artery, RPA: right pulmonary artery, RSA: right subclavian artery, RV: right ventricle, 3R: 3rd rib. Modified and cited with the permission of the authors and publisher from references 26) and 27).
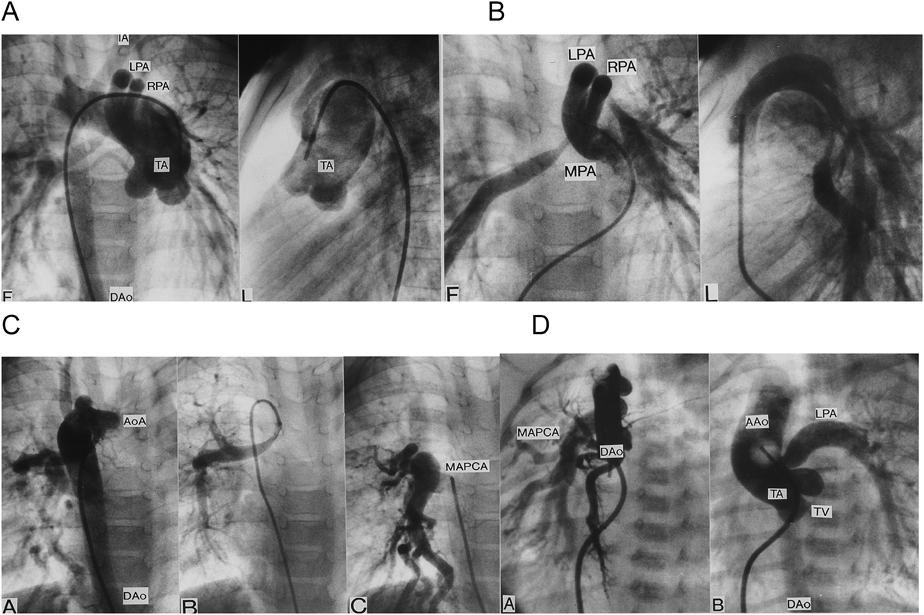
(A–C) TA, RAA, crossing pulmonary arteries, and MAPCA. (D) TA, RAA, and MAPCA. AAo: ascending aorta, AoA: aortic arch, DAo: descending aorta, LPA: left pulmonary artery, MPA: main pulmonary artery, RPA: right pulmonary artery, TA: truncus arteriosus, TV: truncal valve. Modified and cited with the permission of the authors and publisher from reference 29).
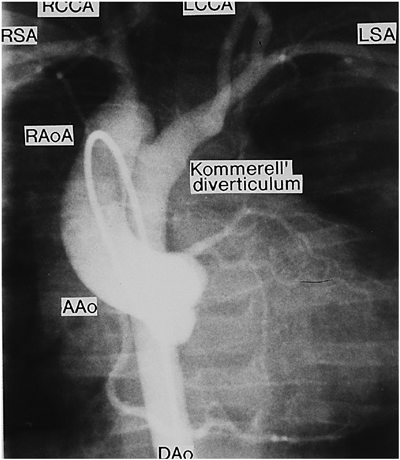
AAo: ascending aorta, DAo: descending aorta, LCCA: left common carotid artery, LSA: left subclavian artery, RSA: right subclavian artery. Vascular ring: Kommerell’s diverticulum to LSA, LCCA, and closed DA.
22q11欠失症のFallot四徴症では右側大動脈弓,鎖骨下動脈レベルに達する高い大動脈弓,鎖骨下動脈起始異常,漏斗部心室中隔全欠損の合併がdel22欠失を合併しないFallot四徴症より多い26, 27).欠失症例での頻度と非欠失例での頻度が次のように報告20)されている.すなわち,右側大動脈弓(70% vs 23%)(欠失例での% vs非欠失例での%),高い鎖骨に届く大動脈弓(50% vs 10%),動脈管欠損+MAPCA(91% vs 50%)などである.漏斗部中隔全欠損のFallot四徴症(Fig. 2A)は欠失症Fallot四徴症では30%,非欠失症Fallot四徴症では5%である.したがってFallot四徴症症例にこれら稀な異常を合併したときには染色体22q11欠失の検索をすべきである31).本症候群のFallot四徴症+肺動脈閉鎖(Fallot四徴症極型)では動脈管開存を合併する例は少なく,大部分がMAPCAを合併する27).
大動脈弓離断は離断部位によりA型,B型,C型の3病型に分類される.A型は大動脈弓の左鎖骨下動脈左で,B型は大動脈弓の左総頸動脈左で,C型は大動脈弓の右総頸動脈左で離断している.本症の大動脈弓離断ではB型になる28).この論文を私が雑誌に投稿したら編集者Anderson教授に人種差があるかもしれないから論文題に〈日本人では〉と入れるよう指示された.不本意ながらそのように加筆して発表したら,この症候群に詳しく,以前から面識のあるローマのMarino教授がローマでも同じだと追加Commentを出してくれた.本症の総動脈幹残遺にはMAPCA,稀な疾患である肺動脈の交差性起始(crossing pulmonary artery)(Fig. 3B)を伴うことがある29).総動脈幹残遺は予後不良のため胎児例の17%に比べて生後例では染色体22q11欠失検索例が少ない.本症で単独に心室中隔欠損を合併することもあり,その場合に大動脈異常(憩室,血管輪)(Fig. 4)を合併することがある30).
高尾教授は円錐動脈幹異常顔貌症候群を1976年に初めて記載してから5年ごとに国際シンポジウムを開催してその成因を追求し,1993年に染色体22q11.2欠失を突き止められた.形態形成の研究は山岸教授が進めており,山岸敬幸,白石公編著の心臓発生学書32)に結実している.高尾教授定年の1993年以降の私達の欠失確定症例の心臓病像研究26–30)は国際的にも注目されて,2001年には米国心臓病学会(American College of Cardiology)総会での先天性心疾患シンポジウムでの門間への指名講演となり,さらに大型の成書である心臓病学書Crawford, DiMarco編集のCardiology(Mosby,2001年)のFallot四徴症と総動脈幹残遺の章10頁を門間が依頼されて執筆した.
高尾教授は高尾シンポジウムのProceeding(1980)の書名をEtiology and Morphogenesis of Congenital Heart Diseaseとされた.また定年退職時に記念に弟子が頂いたお写真(Fig. 1)には,“Development. Unfolding”とメッセージを書かれた.この書名とメッセージから高尾教授の熱い想いが伝わってくる.
利益相反に関する開示事項はありません.
1) Gumbiner CU, Takao A: Ventricular septal defect, in Garson A Jr., Bricker JT, McNamara DG (eds): The Science and Practice of Pediatric Cardiology. Philadelphia, Lea & Febiger, 1990, pp 1002–1022
2) 榊原 仟,広沢弘七郎,高尾篤良,ほか:心研創立四十周年記念誌.東京,東京女子医科大学付属日本心臓血圧研究所,1995,pp 170–175, 260
3) 木内晶子,高尾篤良:円錐総動脈幹異常児の顔貌.(目で見る小児科)小児科 1976; 17: 巻頭2頁
4) Takao A, Ando M, Cho K, et al: Etiologic categorization of common congenital heart disease, in Van Praagh R, Takao A (eds): Etiology and Morphogenesis of Congenital Heart Disease. Mount Kisco, New York, Futura Publishing Co., 1980, pp 253–269
5) DiGeorge AM: Discussions on a new concept of the cellular basis of immunity. J Pediatr 1965; 67: 907–908
6) Shprintzen RJ, Goldberg RB, Lewin ML, et al: A new syndrome involving cleft palate, cardiac anomalies, typical faces, and learning disabilities. Cleft Palate J 1978; 5: 56–62
7) Shimizu T, Takao A, Ando M, et al: Conotruncal anomaly face syndrome: Its heterogeneity and association with thymic involution, in Nora JJ, Takao A (eds): Congenital Heart Disease. Causes and Processes. Mount Kisco, NY, Futura, 1984, pp 29–41
8) de la Chapelle A, Herva R, Koivisto M, et al: A deletion in chromosome 22 can cause DiGeorge syndrome. Hum Genet 1981; 57: 253–256
9) Kelley RI, Zackai EH, Emanuel BS, et al: The association of DiGeorge anomalad with partial monosomy of chromosome 22. J Pediatr 1982; 101: 197–200
10) Scambler PJ, Kelly D, Lindsay E, et al: Velo-cardio-facial syndrome associated with chromosome 22 deletions encompassing DiGeorge locus. Lancet 1992; 339: 1138–1139
11) Driscoll DA, Spinner NB, Budarf ML, et al: Deletions and microdeletions of 22q11.2 in velo-cardio-facial syndrome. Am J Med Genet 1992; 44: 261–268
12) Wilson DI, Cross IE, Goodship JA, et al: DiGeorge syndrome with isolated aortic coarctation and isolated ventricular septal defect in three sibs with a 22q11 deletion of maternal origin. Br Heart J 1991; 66: 308–312
13) Burn J, Takao A, Wilson D, et al: Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22q11. J Med Genet 1993; 30: 822–824
14) Momma K: Cardiovascular anomalies associated with chromosome 22q11.2 deletion. Int J Cardiol 2007; 114: 147–149
15) Matsuoka R, Kimura M, Scambler PJ, et al: Molecular and clinical study of 183 patients with conotruncal anomaly face syndrome. Hum Genet 1998; 103: 70–80
16) 松岡留美子,砂原真理子,古谷道子(編):22q11.2欠失症候群.東京,中山書店,2010, pp 1–159
17) Lindsay EA, Vitelli F, Su H, et al: TBX1 haploinsufficiency in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 2001; 410: 97–101
18) Yagi H, Hurutani Y, Hamada H, et al: Role of TBX1 in human del22q11.2 syndrome. Lancet 2003; 362: 1366–1373
19) Yamagishi H, Srivastava D: Unraveling the genetic and development of 22q11 deletion syndrome. Trends Mol Med 2003; 9: 383–389
20) Momma K: Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome. Am J Cardiol 2010; 105: 1617–1624
21) Boudjemline Y, Fermont L, Le Bidois J, et al: Prevalence of 22q11 deletion in fetuses with conotruncal cardiac defects: A 6 year prospective study. J Pediatr 2001; 138: 520–524
22) Ryan AK, Goodship JA, Wilson DI, et al: Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: A European collaborative study. J Med Genet 1997; 34: 798–804
23) Park I-S, Ko J-K, Kim Y-H, et al: Cardiovascular anomalies in patients with chromosome 22q11.2 deletion: A Korean multicenter study. Int J Cardiol 2006; 114: 230–235
24) Marino B, Digilio MC, Toscano A, et al: Anatomic patterns of conotruncal defects associated with deletion 22q11. Genet Med 2001; 3: 45–48
25) Oskarsdottir S, Person C, Erikson BO, et al: Presenting phenotype in 100 children with 22q11 deletion syndrome. Eur J Pediatr 2005; 164: 146–153
26) Momma K, Kondo C, Ando M, et al: Tetralogy of Fallot associated with chromosome 22q11 deletion. Am J Cardiol 1995; 76: 618–621
27) Momma K, Kondo C, Matsuoka R: Tetralogy of Fallot with pulmonary atresia associated with chromosome 22q11 deletion. J Am Coll Cardiol 1996; 27: 198–202
28) Momma K, Ando M, Matsuoka R, et al: Interruption of the aortic arch associated with deletion of chromosome 22q11 is associated with subarterial and doubly committed ventricular septal defect in Japanese patients. Cardiol Young 1999; 9: 463–467
29) Momma K, Ando M, Matsuoka R: Truncus arteriosus communis associated with chromosome 22q11 deletion. J Am Coll Cardiol 1997; 30: 1067–1071
30) Momma K, Matsuoka R, Takao A: Aortic arch anomalies associated with chromosome 22q11 deletion (CATCH 22). Pediatr Cardiol 1999; 20: 97–102
31) Gomez O, Soveral I, Bennasar M, et al: Accuracy of fetal echocardiography in the differential diagnosis between truncus arteriosus and pulmonary atresia with ventricular septal defect. Fetal Diagn Ther 2016; 39: 90–99
32) 山岸敬幸,白石 公(編):臨床心臓発生学.東京,メジカルビュー社,2007


This page was created on 2016-12-20T16:00:41.079+09:00
This page was last modified on 2017-02-03T18:25:45.100+09:00
このサイトは(株)国際文献社によって運用されています。