小児肺動脈性肺高血圧症:治療の最前線Advanced Therapies for the Pharmacological Treatment of Pediatric Pulmonary Arterial Hypertension
東邦大学医療センター大森病院小児科Department of Pediatrics, Toho University Omori Medical Center ◇ Tokyo, Japan
発行日:2017年7月1日Published: July 1, 2017
小児期発症の肺動脈性肺高血圧症は,特発性および遺伝性肺動脈性肺高血圧症や先天性心疾患による肺高血圧が多く,成人期発症の肺高血圧症とは異なる.また,小児の肺高血圧症では,臨床症状,治療反応性,予後因子などにおいて,成人といくつかの相違点や類似点が存在することがわかってきている.しかし,治療に関しては,前向き研究やランダム化比較試験が少ないため,未だ承認されている薬剤が少なく,成人の肺高血圧治療戦略を参考に,現在も模索されている状況である.その使用経験の報告からは小児患者においても成人と同等の効果が得られることがわかってきたが,小児への治療戦略は未だ確立されていない.成人における治療をそのまま小児患者に置き換えることは難しいため,近年小児の治療戦略に関して新しい提言が報告されはじめてきた.本稿では,小児期発症の特発性,遺伝性肺動脈性肺高血圧症および先天性心疾患に伴う肺高血圧症における未承認薬を含む薬物治療の有効性や安全性,治療戦略に関する新たな知見をまとめて紹介する.
Pulmonary arterial hypertension (PAH) is a crucial determinant of morbidity and mortality in children and adults. It can present at any age from infancy to adulthood. The clinical features of pediatric PAH, however, differ from those of adults, and in children, there is a predominance of idiopathic PAH (IPAH) or that associated with congenital heart disease (CHD). Without appropriate treatment, the median survival rate after diagnosis of IPAH in children is considerably worse than that in adults. Additionally, though PAH related to CHD in most children may resolve after surgical correction, some children may develop an irreversible pulmonary vascular disease. Recent studies of pediatric PAH have highlighted the unique aspects of pathogenesis and challenging treatments in IPAH or PAH associated with CHD. Treatment with new selective pulmonary vasodilators offers hemodynamic and functional improvement in pediatric populations. Survival within the first 5 years following diagnosis has been reported to be between 62% and 90%, and the survival rate has been similar between IPAH and PAH associated with CHD. However, there is currently limited data from randomized controlled trials in children with PAH for evaluating the safety and efficacy of vasodilator therapies, which are approved for adult patients. Therefore, further studies are required for development of specific strategies for treating children with PAH. This review provides a brief overview of recent information regarding current approaches to PAH in children.
Key words: treatment strategy; Eisenmenger syndrome; upfront combination therapy; sequential combination therapy; congenital heart disease
© 2017 特定非営利活動法人日本小児循環器学会© 2017 Japanese Society of Pediatric Cardiology and Cardiac Surgery
肺動脈性肺高血圧(pulmonary arterial hypertension; PAH)は新生児から成人までのすべての年齢層に生じうる病態であるが,その病因は多岐にわたるだけでなく,成人と小児の間では大きく異なる.このため,成人領域における治療目標や治療戦略をそのまま小児患者で置き換えることは困難である.しかし,実際の臨床現場では,成人PAH領域における大規模なランダム化試験によって検証された治療戦略を,小児患者において修正を加えながら試されている現状がある.実際,欧米で推奨される小児PAHにおける治療戦略の多くはexpert opinionによるものであり,エビデンスレベルの高い治療薬は多くない.また,薬剤に関しては,小児PAHでの使用は一部の薬剤を除きほとんどはoff labelであり,その有効性や安全性,薬物血行動態などの知見に乏しいまま,手探りで投与量が設定されている.小児PAHの治療戦略を立てるにあたっては,成人PAHと異なる特性を理解したうえで治療,管理を行う必要がある.本稿では小児PAHで多い特発性肺動脈性肺高血圧症(idiopathic PAH; IPAH),遺伝性肺動脈性肺高血圧症(heritable PAH; HPAH)および先天性心疾患に伴う肺動脈性肺高血圧症(PAH associated with congenital heart disease; APAH-CHD)に対する治療戦略に関する最新の知見を紹介する.
小児PAHの治療アルゴリズムについては,2015年に米国から,2017年にも欧州からも報告されたが,2つの内容はおおむね変わらないものであり,成人PAHのアルゴリズムを参考に治療戦略が決定されている1–3).Fig. 1に示す通り,まず急性肺血管反応性試験(acute vasoreactivity testing; AVT)を行い,反応例かを判別する.反応性に関する判定は成人の定義と異なり,小児における定義がREVEAL(Registry-to-Evaluate-Early-And-Long-term PAH disease management)で提唱されており,1)平均肺動脈圧が20%以上低下,2)心拍出量が不変,増加,または10%以内の低下,3)pulmonary to systemic vascular resistance ratio(PVR/SVR比)が不変または低下,としている4).この定義による反応例は,IPAHおよびHPAHでは30%程度,APAH-CHDでは13%程度存在する4, 5).このAVTでは,100%酸素(酸素マスクで10分投与),epoprostenol持続静注,一酸化窒素(Nitric Oxide; NO)などによる反応性で検討されてきた.NO吸入試験の多くは20 ppmで判定されるが,マスクによる投与では厳密に濃度を維持できず,NOが空気中に拡散されるために気管内挿管を必要とする点でPAH患者の検査としては不向きといえる.Epoprostenolは,2 ng/kg/minから開始し4 ng/kg/minで判定し,最大でも8 ng/kg/minまでの増量にとどめる.Epoprostenol負荷試験は体血管抵抗の低下による低血圧のリスクを伴うため,体血圧をモニタリングながら慎重に行う必要がある.AVTに反応する症例ではカルシウム拮抗薬で治療を開始してもよいが,実際には本邦でカルシウム拮抗薬の単剤治療は行われることは稀である.また,この治療の経過中に増悪する症例が存在するため,慎重な経過観察と迅速な肺血管拡張薬の開始が必要である6).Nifedipine(2~5 mg/kg/日分3),diltiazem(3~5 mg/kg/日分3),amlodipine(2.5~5 mg/kg/日分2)が小児PAHでは投与されるが,verapamilは心室機能への影響が懸念されるため使用されない.また,高い右房圧,低い心拍出量,1歳未満の症例では,カルシウム拮抗薬は避けることが望ましい7).このAVTは,予後予測として有用とする報告もあり,現状ではリスク層別化の指標として使用されている8).
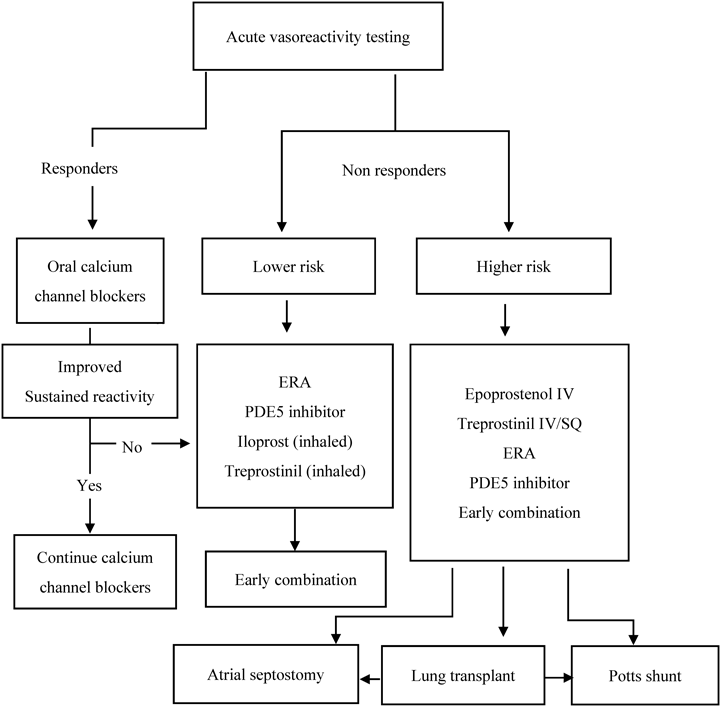
Modified from: Ivy DD, Abman SH, Barst RJ, et al. Pediatric pulmonary hypertension. J Am Coll Cardiol 2013; 62: D117–26. ERA: endothelin receptor antagonist, IV: intravenous, SQ: subcutaneous, PDE: phosphodiesterase. This is a pragmatic treatment algorithm for children with IPAH based on expert opinion.
AVTで反応不良と判断される多くの症例では,選択的肺血管拡張薬が使用される.その際にリスク層別化を行い,低および高リスクの2つに分ける2).Table 1に示す通り,このリスク分類は,成人と異なる血行動態を示すことから,小児PAH症例に対する基準が提唱されている2, 9).成人の分類と大きく異なるリスク因子としては,6分間歩行距離と心肺運動負荷試験の項目が除かれており,MRIによる右心房容積の代わりに心エコーによる右心室の拡張や機能不全が含まれ,肺血行動態ではpulmonary vascular resistance(PVR)indexが追加され,さらに成長発達の項目が付け足されている点などである2, 9).この変更の理由としては,6分間歩行試験や心肺運動負荷試験が年少児では評価が困難であること,小児PAHでは成人に比して右房のcomplianceが良好であるため,重症度と比例して右房圧が上昇しないことや,肺動脈圧よりも肺血管抵抗が重要であることなどが考えられる10, 11).欧米での提言では,低リスク群では内服療法または吸入療法から開始され,高リスク群ではepoprostenol静注またはtreprostinil静注または皮下注から開始し,早期からphosphodiesterase5(PDE5)阻害薬とendothelin拮抗薬との多剤併用療法が考慮される2).
| Lower risk | Determinants of risk | Higher risk |
|---|---|---|
| No | Clinical evidence of RV failure | Yes |
| No | Progression of symptoms | Yes |
| No | Syncope | Yes |
| Growth | Failure to thrive | |
| I and II | WHO functional class | III and IV |
| Minimally elevated | BNP, NTproBNP | Significantly elevated Rising level |
| Echocardiography | Severe RV enlargement/dysfunction | |
| Pericardial effusion | ||
| CI>3.0 L/min/m2 | Hemodynamics | CI<2.5 L/min/m2 |
| mPAP/mSAP<0.75 | mPAP/mSAP>0.75 | |
| AVT positive | RAP>10 mmHg | |
| PVRi>20 WU/m2 | ||
| Table adapted from: Ivy DD, Abman SH, Barst RJ, et al. Pediatric pulmonary hypertension. J Am Coll Cardiol 2013; 62: D117–26. AVT: acute vasoreactivity testing; BNP: brain natriuretic peptide; CPT: cardiopulmonary exercise test; CI: cardiac index; mPAP: mean pulmonary artery pressure; RV: right ventricle, PVRi: pulmonary vascular resistance index; RAP: right atrium pressure; RV: right ventricle; NTproBNP: N-terminal proBNP, mSAP: mean systolic pressure, WHO: World Health Organization. | ||
近年の肺血管拡張薬の進歩によって,成人PAH患者において40%以下であった5年生存率は57%まで上昇しており,IPAHおよびHPAHに限定すると65%程度であることが報告されている12, 13).一方,小児領域ではPAH全体で5年生存率が74%(62~90%)であり,IPAHおよびHPAHで75%,APAH-CHDでも71%と成人PAHに比して予後が良いことがわかってきている(8,14).診断時の肺血行動態は成人と同等からやや不良であることを踏まえると,小児PAH患者が成人よりも肺血管拡張薬への反応性が良好であることを裏づけている8, 12–14).
本邦においても,欧米同様に確立された治療戦略はなく,各施設において様々な治療方針が行われている.我々の施設における治療戦略は,WHO機能分類に基づいた重症度によってリスク層別化を行い,治療薬を選択している.しかし,重症度評価として用いられるWHO機能分類は,一般的に6歳未満の小児患者では評価が困難である.近年,小児用に改変されたものが提唱されており,小児PAH患者における軽度の運動耐応能低下を認める例や失神を伴う例などにおける重症度評価に有用である15).
WHO機能分類がIまたはIIで診断された場合には,経口肺血管拡張薬から開始する.Prostacyclin系の薬剤に併用して,PDE5阻害薬,endothelin拮抗薬の2系統の薬剤のうち,いずれかを選択し,2剤併用のcombination therapyを開始する.忍容性や治療効果を確認しながら経過観察を行い,治療開始後,半年から1年以内に心臓カテーテル検査を行う.治療開始前に比して血行動態の改善がない,または増悪がある場合には,残りの1剤をadd-onし,3剤によるcombination therapyに移行する.ただし,家族歴のあるHPAHの症例では治療介入を積極的に行い,十分な説明のもと患者本人と家族の同意を得て,epoprostenol持続静注療法を初期から行うことも検討される.これは,家族発症の有無にかかわらず,HPAHでは進行性が早く予後不良であることが懸念されるためである.小児HPAHにおけるbone morphogenetic receptor protein 2(BMPR2)とactivin receptor-like kinase(ALK-1)の変異をもつ症例の生存率を前向研究で検討した報告では,いずれかの遺伝子変異のキャリアは非キャリアに比して,5年生存率が不良であり(BMPR2 vs ALK-1 vs非キャリア:55% vs 64% vs 90%),BMPR2変異陽性患者では陰性患者に比して,AVTにおける反応性が不良とする報告もある16, 17).
WHO機能分類IIIの症例では,初期より経口3剤併用の治療で行う.治療開始後,半年から1年以内に心臓カテーテル検査を行い,血行動態の改善度が乏しい場合にはepoprostenol治療を開始する.WHO機能分類IVの症例では,治療前の心臓カテーテル検査が行えないことが多い.臥位になれない最重症例は,集中治療室での管理が望ましい.成人例ではスワン・ガンツカテーテルの挿入によって心房圧と肺動脈圧,心拍出量のモニタリングが可能となるが,特に小児ではカテーテル挿入時の疼痛などによって循環動態が悪化する可能性があり,慎重に行うべきである.我々の施設では,末梢挿入型中心静脈カテーテル(ダブル・ルーメン)を留置し,0.5 ng/kg/min程度のごく少量のepoprostenol持続静注に少量のドブタミン(1~2 µg/kg/min程度)を併用して投与する.同時に動脈ラインを挿入し体血圧をモニタリングする.また,末梢挿入型中心静脈カテーテルで心房圧をモニタリングし,心臓超音波によって三尖弁逆流から肺動脈圧を推定しながら血行動態を確認する.重症度が高い症例ほど体うっ血が強いが,浮腫の改善を目的に過量の利尿剤を投与すると,PAHでは心拍出量は極めて低いために血行動態を増悪させる.輸液量は維持量で投与し,利尿剤は点滴持続静注で少量から開始するほうがよい.Epoprostenol治療に反応があると,心拍出量が増加し徐々に尿量が増加する.数日みて反応が乏しければ0.5 ng/kg/minずつ増量してもよい.その後は輸液量を減量し,体血圧をみながらepoprostenolを1週間から2週間ごとに0.5~1.0 ng/kg/minずつ緩徐に増量していく.急性期には肺動脈圧の低下よりも,心房圧の低下や心拍出量の増加に伴う尿量の増加,末梢循環障害の改善などを治療目標とすべきである.
治療戦略に関して肺血管拡張薬にfocusがあたることが多いが,そのほかの支持療法もPAH治療において重要である.
A)酸素終日または移動時の酸素療法は,労作時息切れの軽減に有用である.しかし,酸素療法の長期使用によって予後の改善が得られたとする報告はない.このため,日常生活における臨床症状や睡眠中の低酸素血症の改善を期待して使用される.
B)利尿剤先に述べたように,furosemideなどの利尿剤の過量投与は心拍出量の低下や末梢循環減少を招く.高度の肺動脈圧上昇を伴う症例ほど,肺循環を維持するためには,ある程度の心房圧が必要であり,静注による利尿剤投与は急激な血行動態の変化を来す可能性があり注意が必要である.Spironolactoneによるrenin angiotensin aldosterone系の抑制は,PAHにおける心不全に対しても有効とする報告がある.成人PAHにおけるambrisentanの治療効果を検討したARIES(ambrisentan in pulmonary arterial hypertension randomized, double-blind, placebo-controlled, multicenter efficacy)trialsでは,ambrisetan単独よりもspironolactone併用群のほうが予後良好であった18).この結果からspironolactoneはPAH症例においても,利尿作用以外に抗心不全作用としての別の働きがあることが示唆される19).
C)抗凝固療法成人PAHにおけるwarfarinの長期投与が予後を改善させた報告はあるが,小児での検証は未だない20).New oral anticoagulantsも同様に小児での報告はない.成人PAHのガイドラインではIPAH, HPAHに対して推奨されているが,小児PAHにおける抗凝固療法の使用に関する明確な記載はない21).しかし,成人同様に小児PAHでも主肺動脈の拡張を来し,肺動脈内血栓を伴うことがあり,抗凝固療法は必要と考えられる22).しかし,喀血を認めた症例では中止が必要であり,鼻出血を伴うALK-1遺伝子変異をもつ症例では回避されることがある.また,epoprostenolをはじめ血小板凝集に影響する薬剤を投与されている症例では潜在的な出血のリスクが伴っており,warfarin治療においてはInternational normalized ratioは1.5前後を目標とし,2.5を超える場合には減量が望ましい23).
Nice分類ではAPAH-CHDを大きく4つのカテゴリーに分けている1).I群はEisenmenger syndrome(ES)例,II群はPVRの上昇を伴い,かつ安静時の酸素飽和度は正常である,未手術の左–右短絡性疾患,III群は,本来PAHを来さない小さな短絡(co-incidental)を伴うIPAHと似た血行動態を示すもの,IV群はCHD術後に残存,または再発するPAHに分類されている.
ESは高肺血流量によって肺血管におけるリモデリングが起こり,結果的に肺動脈圧とPVRが上昇するため,欠損孔を介した右–左性短絡を来し,常時チアノーゼを呈する状態を指す.低酸素血症による運動耐容能の低下のみならず,二次性赤血球増多症と過粘稠度症候群,喀血,血栓塞栓症,不整脈,腎機能低下などの全身性疾患を合併し,進行する右室機能不全を伴う症候群である.主要となるCHDでは,post-tricuspid shuntである心室中隔欠損や動脈管開存症が肺血管閉塞性病変を来しやすく,pre-tricuspid shuntの代表である心房中隔欠損では稀とされている.しかし心房中隔欠損においても,加齢に伴いES化する一群が存在しており,およそ20%前後において肺血管のリモデリングを生じることが報告されている24).
ESの治療戦略としては,IPAHに準じた治療薬が記載されているが,エビデンスに基づくものはほとんどない21).その中でも,ES患者に対するBREATHE-5(Bosentan Randomized Trial of Endothelin Antagonist Therapy-5)では,プラセボ群に比してbosentan治療群が,PVRや運動耐容能を改善させたことを報告している25).Sildenafil, ambrisentan, epoprostenol治療も,運動耐容能,酸素飽和度,WHO機能分類,PVRなどを改善させたとする報告もあるが,いずれも少数のopen label試験であり,その効果に関してはまだ確立されたものではない26, 27).
この群においては,短絡性疾患によるPAHであることより,修復術が根本的な治療となるが,PVRの上昇を伴う症例では,手術の可否について検討する必要がある.治療アルゴリズムでは,一般的には,PVR indexが6 WU·m2以下であるかによって手術適応を判断する(Fig. 2)28, 29).PVR indexが6~8 WU·m2のborderlineの症例についは施設ごとの判断に委ねられているが,術後のPAH残存のリスクが高い症例ではAVTにおける反応性を確認する.この際の基準はIPAHとは異なり,PVR index<6 WU·m2かつPVR/SVR<0.3の基準となっており,これに加え,肺体血流比が1.3~1.5程度まで上昇すれば手術は可能,と判断してよい.しかし,AVTに反応があった場合でも,閉鎖術を行った後のPAH残存が懸念される.Repair and treatの治療方針として,術後に肺血管拡張薬を投与しながら心臓カテーテルによる再評価を行うことが望ましい.
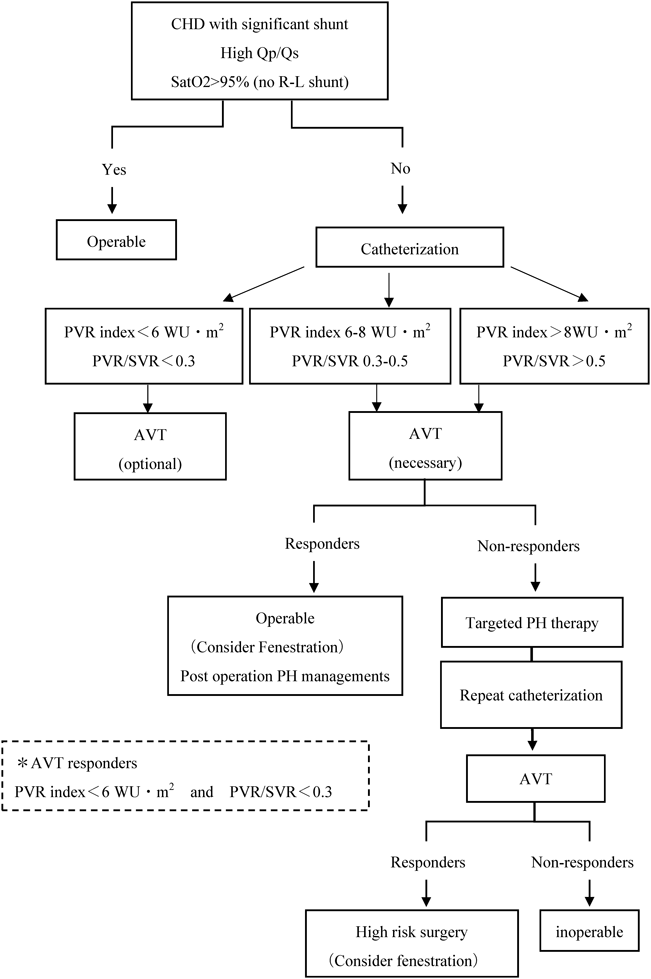
Modified form Ref. 28) Kozlik-Feldmann R, Hansmann G, Bonnet D et al: Pulmonary hypertension in children with congenital heart disease (PAH-CHD, PPHVD-CHD): Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 2016; 102 Suppl 2: ii42–ii48. AVT: acute vasoreactivity testing, CHD: congenital heart disease, PVR: pulmonary vascular resistance, PVR/SVR: pulmonary to systemic vascular resistance ratio, Qp/Qs: pulmonary to systemic blood flow ratio, Sat O2: oxygen saturation. Indications for invasive diagnostic procedures and eligibility for surgery (comprehensive heart catheterization) includes basic evaluation and acute vasoreactivity testing.
PVR indexが8 WU·m2を超える症例は,原則修復術は禁忌と考えられるが,近年では肺血管拡張薬を術前,術後に投与することにより,PVRの高い症例に対しても欠損孔の閉鎖を行う“Treat and repair, Repair and treat”という治療戦略が試みられるようになっている30, 31).しかし,欠損孔があるESと比較した長期的予後については未だ不明であり,十分なエビデンスがあるわけではない.また,短絡性心疾患に対するTreat and repairなどの使用経験に関しては症例報告に留まり,どの薬剤が推奨されるかの検討はなされていない.
CHDによる高肺血流の時期が過去に認めなかった小さな短絡であったにもかかわらず,高度のPAHを呈した症例が該当する.IPAHにCHDが偶然合併した症例とも考えられるが,重症度は決して低くはない.II群とは異なり,短絡の閉鎖術による血行動態の是正を行うことがPAHを改善させることはなく,閉鎖によって血行動態を増悪させるため外科的介入は禁忌である.治療戦略は原則IPAHの症例と同様である.
欠損孔の修復術後に有意な遺残短絡がないにもかかわらず,術後早期から遠隔期にかけてPAHが再燃,進行する.心室中隔欠損,動脈管開存,完全型房室中隔欠損などの単純型の短絡性疾患以外にも,総動脈幹症,大動脈肺動脈窓,完全大血管転位などの術後で生じることが知られている.血行動態としてはIPAHと同じであり,使用される肺血管拡張薬も同様である.しかし,薬剤治療への反応性や予後は,遺残短絡のあるPAHやIPAHに比して不良な症例も多く,より積極的な治療介入を行う必要がある.
4つのカテゴリーのいずれにも含まれない群である.近年ではFontan型手術後において,平均肺動脈圧が25 mmHgを下回る場合でもPVRが3 WU·m2を超えるか,transpulmonary gradient(平均肺動脈圧—肺動脈楔入圧)が6 mmHgを超える場合にはPAHと定義するようになってきた32).肺動脈圧が低いにもかかわらず予後不良であり,この疾患群におけるPAHについては議論が必要である.治療戦略に関しては,Fontan型手術の周術期における肺高血圧に対しNO吸入療法とepoprostenol治療で肺血行動態を改善した報告や,術後遠隔期における肺高血圧に対してmacitentan(成人10 mg/日1日1回)とbosentan(小児2 mg/kg 1日2回)治療が奏功し,運動耐容能の改善が得られた報告などがあるが,いずれも少数例の報告である33–35).また,Fontan型手術後の肝機能障害の報告は多く,bosentanの使用に関しては注意が必要である.
小児PAH患者における予後もここ10年間,治療の進歩により劇的に改善してきた.しかし,ほとんどの治療薬が,小児では未だoff labelとして使用されており薬物動態に関するデータも少ないことから,用量設定においてもコンセンサスが得られたものは少ない.各製剤の比較をTable 2に示す.
| ||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
Epoprostenol(フローラン®,エポプロステノールACT®)は持続静注により投与する.小児での承認が得られているのは,エポプロステノールACT®である.Epoprostenolは,他のPAH治療薬で十分な効果を得られないWHO機能分類IIIまたはIVのPAHで投与される.低用量(1~2 ng/kg/min)から開始し,副作用や忍容性に注意しながら1か月ごとに1 ng/kg/minずつ増量していく.開始後2~3年で維持量(20~30 ng/kg/min)に到達する.IPAH患者へのepoprostenolの効果は用量依存性があると考えられており,投与開始1か月までに10~20 ng/kg/min程度まで短期間に用量を増やす方法や,平均肺動脈圧が40 mmHg以下を目標に100 ng/kg/min以上まで増量していく方法も試されているが,副作用も用量依存性に出現するため,忍容性が低くなる可能性がある.現在,増量方法に関しては各施設から様々な治療方針が提唱されているが,高い肺動脈圧の割に比較的心拍出量が保たれている小児患者に対しepoprostenol増量により,肺動脈圧は下がらないものの心拍出量だけが増加し,動悸や静脈還流量の増加による体液貯留をきたす可能性がある.また,治療反応性も各症例で異なるため肺血行動態や副作用をモニタリングしながら,より慎重な増量が必要である.
おもな副作用に甲状腺機能異常(機能亢進症および低下症のいずれも起こりうる)がある36–38).甲状腺機能亢進に関しては,その発症頻度が小児におけるGrave’s diseaseの発症頻度(1 : 10,000)に比して,小児PAH症例では明らかに高頻度(4~44%)に発症することが報告され,prostacyclinによる副作用の機序が推測されている36–38).Epoprostenol自体が,thyroid stimulating hormone(TSH)を介さずcAMP依存性に直接的に甲状腺ホルモン分泌を亢進させる報告があり,“thyrotropic prostaglandin”の概念が提唱されている39).一方で甲状腺自己抗体を伴うPAH症例があり,自己免疫機序に伴う甲状腺機能亢進も考えられている38, 40).このほかに,BMPR2 mutationが,甲状腺疾患を来しやすいという報告もあり,multifactorialな要因で起きていることがわかってきた41).このため,定期的なTSH, triiodothyronine(FT3),thyroxine(FT4)の値をモニタリングしTSHが低値を示すようであれば,甲状腺自己抗体の測定を行う.比較的病状の安定していたPAH患者が頻脈と心不全症状の増悪を認めた場合には,甲状腺機能亢進の可能性があり,epoprostenol製剤を使用している場合には特に注意が必要である.確立された治療方針はないが,epoprostenol増量中であればいったん増量を中止し,甲状腺機能亢進に対しては抗甲状腺薬,ヨウ化カリウム製剤などでFT4やTSH値が正常化するように投与を行う.治療によっても繰り返される甲状腺機能亢進,甲状腺クリーゼ,抗甲状腺薬の副作用などでコントロールが難しい場合には,甲状腺摘出を検討する36).外科治療に際し,気管内挿管や周術期の甲状腺ホルモンの変動によって急性増悪する可能性があり,経験の多い施設での治療が望ましい.
Epoprostenolの減量または中止については,治療の奏功による離脱症例が報告されてきている.Ivyらの報告では,epoprostenol治療を行った(中央値7年間,2~10年間),8例の小児IPAH症例(中央値12歳,8~17歳)でbosentanを併用し,epoprostenolを減量することができ,3例で血行動態の増悪なくepoprostenolの中止が可能であった42).しかし,成人の報告ではepoprostenolから,bosentanまたはsildenafilへ計画的に移行を行った13例のうち,4例が血行動態および臨床的な増悪を認め,2例がepoprostenol治療を再開している43).このような結果からも,確立された離脱可能な基準や方法はないため,Epoprostenol治療によって正常肺動脈圧に近い値まで低下するような,劇的な血行動態の改善を認めた症例においてのみ限定されるべきである.
半減期が4.5時間と長く,室温で管理できる利点がある.Epoprostenolからtreprostinilへの移行を行った検討が,成人および小児PAH症例で報告されている44–47).Sitbonらの報告では,12例の成人PAHでWHO機能分類IまたはIIの比較的安定した症例で,epoprostenolからtreprostinilの持続静注に1 : 1の用量に切り替えたのち,12週間かけてepoprostenol(28 ng/kg/min)の2倍量の用量までtreprostinil(60 ng/kg/min)を増量した46).この移行方法では臨床的増悪や有害事象なく全例で成功している.小児の報告では,Ivyらが,WHO機能分類I~IIIの臨床的に比較的安定している13例のPAH症例でepoprostenolからtreprostinilに移行した45).この報告では,2通り(slow, rapid)の方法で移行を行っている.Slow transitionでは,epoprostenolと同量のtreprostinil開始後,epoprostenolを半量に減量する.その後1~2時間ごとにepoprostenolを10 ng/kg/minずつ減量し,treprostinilを5~10 ng/kg/minずつ増量する.Rapid transitionは,epoprostenolを中止し同量のtreprostinilを開始する.その後20分~1時間ごとにtreprostinilを5~10 ng/kg/minずつ増量する方法である.いずれの方法でもtreprostinilの目標用量はepoprostenolの1.25~1.75倍としている.全例で移行は成功しており,これに伴った臨床的増悪はなかった.本邦での症例報告では,epoprostenol投与中に血小板減少を示した症例でepoprostenolからのtreprostinilへ移行され,血小板数の回復が得られている.過去の報告にならい,薬剤変更に際しepoprostenolの持続投与量よりも,treprostinilは増量が必要とされ,120~150%の幅で持続投与量を増やす.しかし,実際にはそれより高用量(~200%)が必要になるような症例も存在し,適正な用量設定は事実上困難である.重要なのは,移行の方法や用量設定ではなく,十分選択された患者に対して行われるべきことである.このため,入院治療を要するような重症PAHの症例においては,この薬剤変更によって病状の悪化の可能性があるため推奨されない.
皮下注射のtreprostinilの治療効果は,成人PAHにおけるプラセボ対照二重盲検比較試験で比較的良好な結果が報告されている48).問題となる注射部位の痛みに関しては,鎮痛剤の使用などを行って比較的良くコントロールされている.Treprostinil皮下注の長期成績をみた報告では,6か月以上使用に耐えられた症例ではその後継続投与が可能であった49).
吸入治療薬であるIloprostでは体血管への影響が少なく,より肺血管選択性が高いが,1日6~9回の吸入回数が必要な点や,小児では特に気道過敏性の亢進による咳嗽の頻度が多いため,小児PAH患者における実効性については難しい点が多い.本邦では未販売の吸入用treprostinilのその長い半減期のため,吸入回数が1日4回以下で使用できることは利点といえる.吸入用のtreprostinilの小児PAH患者に対する安全性と有効性が後方視的研究で報告されている50).この検討では,小児PAH症例(29例)に対し,3~9吸入/回(6 µg/1吸入)を1日4回施行し,6週間以上投与した.WHO機能分類は19例で改善,6分間歩行距離も治療前後で改善傾向を示したが,4例で気道攣縮やPAH症状の増悪のため中止しており,15例では中止は至らなかったものの咳嗽や咽頭痛などの症状の訴えがあった.現在,小児PAH患者に対する吸入治療薬は,軽症例または他のPAH治療薬への追加療法としての位置づけとして考えられている.
Sildenafilは欧州では小児PAHに承認されているが,米国と本邦では未だ承認されていない.2012年にsildenafilの有効性および安全性を評価したプラセボ対照二重盲検比較試験であるSTART-1 trail(The Sildenafil in Treatment-Naive Children, Aged 1 to 17 Years, With PAH)が行われた51).その中で,肺血行動態,運動耐応能およびWHO機能分類を改善させることが報告された.その追跡研究として,sildenafil投与における長期予後を検討したSATRT-2 trailが報告された52).235人のPAH患者(IPAHおよびAPAH-CHD,体重>8 kg)を低用量,中等量,高用量の3つの群に分け3年以上の長期予後を検討したところ,3年生存率は低用量から高用量までそれぞれ,94%,93%,88%であった.この結果から,sildenafilは小児PAH患者において長期予後を改善させる可能性が示唆されたが,高用量のsildenafilが低用量と中等量に比して予後がやや悪い結果であった.この原因として,sildenafil高用量群において,低用量群に比してIPAH症例が多く,検討開始時の平均肺動脈圧,PVR indexが高値であり,また,死亡した症例の40%がWHO機能分類IIIまたはIVであった.このため,sildenafilの用量が死亡の直接的な要因になっている可能性は低いと考えられるが,1~17歳における小児PAHに高用量のsildenafil(1回投与量20 mg; <20 kg 40 mg; 20~45 kg 80 mg; >45 kg, 1日3回投与)投与に関しては,死亡のリスクが高くなる可能性があるために,避けるべきとの見解になっている.
一方,tadalafilは長時間作用型のPDE5阻害薬であり,1日1回投与が可能である.成人PAHに対するプラセボ対照二重盲検比較試験では,運動耐応能などを改善することが報告されている53).小児患者におけるランダム化比較試験は未だ行われていないが,少数例の小児PAH(33例)への使用経験が報告されている54).対象患者のうち29例は,sildenafilからtadalafilへ移行した症例であり,残り4例がPED5阻害薬治療に対しde novo症例である.Sildenafil投与量は3.4±1.1 mg/kg/day,移行したtadalafil投与量は1.0±0.4 mg/kg/dayであり,tadalafilへ変更の後から発現した副作用や症状の増悪で中止になった症例はいなかった.また,興味深い点として,sildenafilから移行後に肺血行動態が有意に改善を認めた.これはtadalafilによる内服コンプライアンスの改善や,安定した血中濃度を維持できることが影響していた可能性があり,小児PAH患者に対するtadalafil治療の優位性が示唆された結果であった.また,小児PAH患者(生後3か月~17歳)におけるtadalafilの薬物動態の検討が行われており,0.97 mg/kg/dayの投与量で十分な有効血中濃度が得られ,bosentan併用下においても影響を受けないことが報告されている55).同様に,小児PAH患者においてtadalafilとsildenafilの間で副作用発現に差はなく,tadalafilがsildenafilに比して運動耐応能の改善することも報告されている56).
Riociguatは,血管内皮障害によってNO産生が低下している重症PAH患者に対し,可溶性グアニル酸シクラーゼを直接刺激し,cGMP産生の促進を通して肺血管拡張能を示す.肺動脈血栓内膜摘除術が不可能な慢性血栓塞栓性肺高血圧に対して,Riociguat治療を行ったCHEST(Chronic Thromboembolic Pulmonary Hypertension Soluble Guanylate Cyclase-Stimulator)trialで有効性,安全性が示された57).ほぼ同時期に報告されたPATENT(Pulmonary Arterial Hypertension Soluble Guanylate Cyclase–Stimulator)trialでは,1日7.5 mgのriociguatをIPAH, APAH-CHDなどの患者群に対して投与され,短期(12週)と長期(1年目)の検討で,運動耐容能と肺血行動態が改善している58, 59).これらの検討は成人に対する治療効果であり,小児PAHへの検討はまだ行われていない.Sildenafilを既に投与されているPAH患者に対するriociguatの併用療法の治療効果を検討したPATENT plus trialでは,肺血行動態に対する追加効果はなく,extension studyでは長期投与しえた症例の41%が低血圧によって投与が中断を要したことから,PDE5阻害薬との併用療法は事実上禁忌とされている60).Sildenafilやtadalafilからriociguatへの移行は可能であるが,いったんPDE5阻害薬の中止が必要である.その半減期からsildenafilは中止した翌日以降,tadalafilでは2日後にriociguatを開始することが望ましいが,明確に規定されたものはなく,最大3日間のwashout期間を設ければ問題ないと考えられる.また,riociguatは低用量から増量する必要があり,治療域に達するまで時間を要するため,血行動態が不安定な重症患者においては推奨されない.
Bosentanは,2015年に小児PAH患者に対し本邦で初めて承認を得た.2歳から12歳までのIPAHおよびHPAH患者を対象としたFUTURE-1(Formulation of bosentan in pulmonary arterial hypertension)およびFUTURE-2 studyで,bosentanの薬物血行動態と臨床的有効性と安全性が示された61, 62).FUTURE-1では,2歳から11歳までのIPAHおよびHPAHに対して施行され,bosentanを2 mg/kgまたは4 mg/kgを1日2回の2つの治療群で比較検討したところ,両群で薬物動態はほぼ類似しており,WHO機能分類の改善を認めた.Bosentanによる肝機能異常は12週目の時点では認めず,約2年間の経過観察をしたFUTURE-2でも33例中1例のみであった.成人に比して肝機能障害の頻度が年少児ほど少ないため,小児での忍容性は高い可能性が示唆される.また,ESを含むAPAH-CHDに対するbosentanの有効性は多くの検討で証明されており,最近ではTEMPO(Treatment With Endothelin Receptor Antagonist in Fontan Patients, a Randomized, Placebo-Controlled, Double-Blind Study Measuring Peak Oxygen Consumption)studyで,Fontan術後の若年成人においてbosentanによる運動耐容能の改善が示されている63).
Ambrisentanはendothelin A受容体に選択的に作用し1日1回投与が可能であり,小児PAHでの使用経験と薬物動態が報告されている64).対象患者のPAH38例のうち,15例はbosentanからのtransitionであり,残り23例は病状の進行に対しadd-on治療として投与されている.Transitionおよびadd-onともに,有意に肺血行動態は改善しており,およそ1/3の症例でWHO機能分類は改善していた.経過観察中,5例(13%)ではambrisentanの副作用としての頭痛,失神および治療効果に乏しい,などの理由で中止されていたが,重篤な肝機能障害での中止例は認めなかった.
Macitentanは,bosentan同様に非選択的エンドセリン受容体拮抗薬であり,小児ではoff labelである.他の2剤に比して受容体解離動態が緩徐で,結合性が高いこと,組織移行性が良好な点で,優れた効果を示すことが期待される薬剤である.Ambirisentanと同様にbosentanに比して肝機能障害の頻度は低く,sildenafilとの相互作用はない.成人PAH患者に対するSERAPHIN試験(Study with Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome)では,肺血行動態,運動耐容能,WHO機能分類を改善させ,10 mg 1日1回の投与法となっている65).小児での投与量は不詳である.
心房間短絡のあるPAH症例は短絡のない症例に比して予後が良いことから発想された治療法である.これは,短絡のないIPAHよりも,右左短絡の存在によって圧負荷による右心不全の進行が緩徐なためである.Atrial septostomyは,繰り返す失神合併例や進行性の右心不全を示す症例が対象となるが,その臨床効果や安全性については報告によって様々であり,一定した効果が期待されるわけではない.術後の右–左短絡による強い低酸素血症を来たすため全身麻酔下で行われるべきであるが,気管内挿管自体のリスクを伴い,術中死亡の可能性も十分考えられる.また,心房間交通が15~17%の症例で術後に閉鎖することが成人PAHで報告されている66, 67).移植までのbridge therapyとして行われたseptostomyが,標準的治療に比して予後を改善させたとする報告もあり,最大限の内科的治療によっても病状の進行を認める症例に限定して行われるべきであろう66–68).小児PAHの20例(施行時の平均年齢8歳)に対してseptostomyの報告例がある69).平均7 mmの欠損孔を作成し,1例で高度の酸素飽和度の低下を示した症例を除いた19例で成功し,術中死亡は認めなかった.酸素飽和度は7.8%低下したが,失神を含む臨床症状の改善や右心機能の改善を認め,術後2年目で18例が生存していた.
Potts短絡術は,左肺動脈と下行大動脈の間に直接シャントを造設するもので,チアノーゼ性心疾患に対して一時期行われてきた姑息的外科治療である70).この手術により,IPAH患者を動脈管開存症に伴うESと同じ血行動態に変えることで,右心不全の進行を軽減させることを目的としている.小児IPAHの8例(2~17歳)に対するPotts短絡術の長期成績が報告されている71).全例WHO機能分類IVであり,内科治療でも改善が認められない患者が対象となっている.術後に下肢の酸素飽和度が平均83%まで低下しており,平均ヘマトクリット値は38~53%まで上昇し多血症の傾向を示した.2例は術後の肺高血圧クリーゼで亡くなっているが,残り6例では6分間歩行距離は改善し,WHO機能分類はIまたはIIへと著明な改善を認めている.平均5年間の経過観察で,短絡部位の狭窄によって再手術を要した症例は認めなかった.重症PAHに対する経カテーテルPotts短絡形成術による報告では,施行された4例のうち2例は治療後死亡したが,残り2例は症状の改善を認めている72).本検討は少数例の報告であり,多くの検討が必要であるが,内科治療に抵抗性でWHO機能分類IVの症例に対する新たな治療のオプションとなる可能性がある.
PAH治療薬のprostacyclin系,NO系,endothelin系の3系統が使用されるようになった2000年初頭から,ある治療目標に向かって治療のエスカレーションを行うgoal oriented therapyが提唱された73).この治療戦略は,運動耐容能の改善や平均肺動脈圧の低下を目標として治療を追加していくものであり,その結果予後が改善するということが報告された.この概念に基づき,各系統のPAH治療薬をまず単剤で投与して効果を確認し,治療目標に達せられない時には他剤を追加するsequential combination therapyが治療の主流として行われてきた.
その後,monotherapyよりcombination therapyが予後を改善させることや,不十分な初期治療が最終的な予後を不良にする可能性がある報告などから,より早期から3系統のうち2つ以上の治療を同時に開始することが有効であるとする治療戦略が提唱された74, 75).2014年にフランスから,IPAHに対して3系統の肺血管拡張薬をほぼ同時に開始し,血行動態と予後の改善を認めた報告がなされた74).その後,前向き二重盲検試験によるambrisentanとtadalafilのupfront combination therapyが発表された75).この論文で,同時併用療法が各々の単独療法よりも運動耐容能や予後を改善させることが報告された.Upfront combination therapyのコンセプトとしては,重症PAH患者に対し一定の目標に向かって治療内容を修正していく方針では介入の遅れにつながるため,治療目標よりも最終的な予後を改善させることを目的に,最初から積極的治療介入を行ったほうがよい,というものである.もちろん,複数の系統の薬剤を同時に使用することによって,より薬剤の副作用が問題となることは当然予想される.予定された投与量に達することが困難になる可能性や,忍容性の低下や治療の中断のリスクも十分考えうるが,現在ではこのupfront combination therapyが予後改善につながる,より効果的な治療戦略であると考えられている.
しかし,小児のPAH患者においては,この治療戦略が果たしてsequential combination therapyを上回るかは未だ不明である.成人のPAH患者と異なり小児では,高い肺動脈圧を呈する割に心拍出量が保たれ治療反応性が比較的良好であることを考えると,同時併用治療による治療効果が成人患者ほど有意な差として現れるかは疑問である.また一方で,治療目標となる指標を持たないこの治療戦略が,結果的に期待される結果をもたらさなかった場合に,次の治療方針を何に向かって計画していくかが不透明である.従来のsequential combination therapyによっても10年以上生存している症例が存在している現状において,重症かつ進行性の疾患であるPAHに対し,このupfront combination therapyが5年以内の予後を改善させたからといって,その後10年以上の予後を保証しているわけではない.リスク層別化のない治療戦略が,期待された効果が得られず行き詰った場合のレスキューとなる治療を提示できなければ,長期的な予後の改善につながるか難しくなる可能性がある.さらに,upfront combination therapyがPAHの診断がなされた全例で本当に必要なのかは現時点では不明であり,患者の血行動態などからリスク層別化を行い,この治療戦略の恩恵を受けることができる症例を選別することが,今後必要となってくるのかもしれない.
小児期発症PAHは,非特異的な臨床症状を認めるため,診断に至るまで時間を要することがある点や進行が早いことなどから,従来予後不良と考えられてきた.しかし,近年PAHに関する知見が深まり,より早期に診断がなされるようになったこと,小児PAHは肺血管拡張薬に対する反応性が比較的良好であることから,生命予後の改善が得られるようになってきた.しかし,未だに欧米においても小児患者への肺高血圧治療薬に対する大規模な臨床研究が行われていない.本邦でも患者の絶対数が少ない上,各施設で治療がなされるため,expert opinionによって治療薬が選択されている状態が続いている.今後は,小児PAH患者における各肺血管拡張薬の国際共同試験による治験を進めていき,エビデンスの蓄積ともに小児への適応拡大が成されていくことが期待される.
本論文を投稿中に,共著者である佐地勉名誉教授がご逝去されました.長年に亘るご指導を感謝申し上げると共に,共著者一同,佐地先生のご冥福を心からお祈り致します.
All authors did not receive any honorariums, grants, or other form of payments for this research and have no conflicts of interest to disclose.
1) Simonneau G, Gatzoulis MA, Adatia I, et al: Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62 Suppl: D34–D41
2) Abman SH, Hansmann G, Archer SL, et al: American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Surgery and Anesthesia; and the American Thoracic Society: Pediatric Pulmonary Hypertension: Guidelines from the American Heart Association and American Thoracic Society. Circulation 2015; 132: 2037–2099
3) Hansmann G, Apitz C: Treatment of children with pulmonary hypertension: Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 2016; 102 Suppl 2: ii67–ii85
4) Douwes JM, van Loon RL, Hoendermis ES, et al: Acute pulmonary vasodilator response in paediatric and adult pulmonary arterial hypertension: Occurrence and prognostic value when comparing three response criteria. Eur Heart J 2011; 32: 3137–3146
5) Douwes JM, Humpl T, Bonnet D, et al: TOPP Investigators: Acute Vasodilator Response in Pediatric Pulmonary Arterial Hypertension: Current clinical practice from the TOPP registry. J Am Coll Cardiol 2016; 67: 1312–1323
6) Yung D, Widlitz AC, Rosenzweig EB, et al: Outcomes in children with idiopathic pulmonary arterial hypertension. Circulation 2004; 110: 660–665
7) Vorhies EE, Ivy DD: Drug treatment of pulmonary hypertension in children. Paediatr Drugs 2014; 16: 43–65
8) Zijlstra WM, Douwes JM, Rosenzweig EB, et al: Survival differences in pediatric pulmonary arterial hypertension: Clues to a better understanding of outcome and optimal treatment strategies. J Am Coll Cardiol 2014; 63: 2159–2169
9) Hoeper MM, McLaughlin VV, Dalaan AM, et al: Treatment of pulmonary hypertension. Lancet Respir Med 2016; 4: 323–336
10) Sandoval J, Bauerle O, Gomez A, et al: Primary pulmonary hypertension in children: Clinical characterization and survival. J Am Coll Cardiol 1995; 25: 466–474
11) Rosenzweig EB, Ivy DD, Widlitz A, et al: Effects of long-term bosentan in children with pulmonary arterial hypertension. J Am Coll Cardiol 2005; 46: 697–704
12) Benza RL, Miller DP, Barst RJ, et al: An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012; 142: 448–456
13) Hopkins WE, Ochoa LL, Richardson GW, et al: Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or Eisenmenger syndrome. J Heart Lung Transplant 1996; 15: 100–105
14) Barst RJ, McGoon MD, Elliott CG, et al: Survival in childhood pulmonary arterial hypertension: Insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation 2012; 125: 113–122
15) Lammers AE, Adatia I, Cerro MJ, et al: Functional classification of pulmonary hypertension in children: Report from the PVRI pediatric taskforce, Panama 2011. Pulm Circ 2011; 1: 280–285
16) Chida A, Shintani M, Yagi H, et al: Outcomes of childhood pulmonary arterial hypertension in BMPR2 and ALK1 mutation carriers. Am J Cardiol 2012; 110: 586–593
17) Rosenzweig EB, Morse JH, Knowles JA, et al: Clinical implications of determining BMPR2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J Heart Lung Transplant 2008; 27: 668–674
18) Maron BA, Waxman AB, Opotowsky AR, et al: Effectiveness of spironolactone plus ambrisentan for treatment of pulmonary arterial hypertension (from the [ARIES] study 1 and 2 trials). Am J Cardiol 2013; 112: 720–725
19) Juurlink DN, Mamdani MM, Lee DS, et al: Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351: 543–551
20) Preston IR, Roberts KE, Miller DP, et al: Effect of warfarin treatment on survival of patients with pulmonary arterial hypertension (PAH) in the registry to evaluate early and long-term PAH disease management (REVEAL). Circulation 2015; 132: 2403–2411
21) Galiè N, Humbert M, Vachiery JL, et al: 2015 ESC/ERSGuidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016; 37: 67–119
22) Żyłkowska J, Kurzyna M, Florczyk M, et al: Pulmonary artery dilatation correlates with the risk of unexpected death in chronic arterial or thromboembolic pulmonary hypertension. Chest 2012; 142: 1406–1416
23) McLaughlin VV, Archer SL, Badesch DB, et al: ACCF/AHA: ACCF/AHA 2009 expert consensus document on pulmonary hypertension: A report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: Developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 2009; 119: 2250–2294
24) Steele PM, Fuster V, Cohen M, et al: Isolated atrial septal defect with pulmonary vascular obstructive disease–long-term follow-up and prediction of outcome after surgical correction. Circulation 1987; 76: 1037–1042
25) Galiè N, Beghetti M, Gatzoulis MA, et al: Bosentan randomized trial of endothelin antagonist therapy-5 (BREATHE-5) investigators: Bosentan therapy in patients with Eisenmenger syndrome: A multicenter, double-blind, randomized, placebo-controlled study. Circulation 2006; 114: 48–54
26) Zuckerman WA, Leaderer D, Rowan CA, et al: Ambrisentan for pulmonary arterial hypertension due to congenital heart disease. J Am Coll Cardiol 2011; 107: 1381–1385
27) Rosenzweig EB, Kerstein D, Barst RJ: Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation 1999; 99: 1858–1865
28) Kozlik-Feldmann R, Hansmann G, Bonnet D, et al: Pulmonary hypertension in children with congenital heart disease (PAH-CHD, PPHVD-CHD). Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 2016; 102 Suppl 2: ii42–ii48
29) Lopes AA, Barst RJ, Haworth SG, et al: Repair of congenital heart disease with associated pulmonary hypertension in children: What are the minimal investigative procedures? Consensus statement from the Congenital Heart Disease and Paediatric Task Forces, Pulmonary Vascular Research Institute (PVRI). Pulm Circ 2014; 4: 330–341
30) Kijima Y, Akagi T, Takaya Y, et al: Treat and repair strategy in patients with atrial septal defect and significant pulmonary arterial hypertension. Circ J 2016; 80: 227–234
31) Hu Z, Xie B, Zhai X, et al: Mid term results of “treat and repair” for adults with non-restrictive ventricular septal defect and severe pulmonary hypertension. J Thorac Dis 2015; 7: 1165–1173
32) Cerro MJ, Abman S, Diaz G, et al: A consensus approach to the classification of pediatric pulmonary hypertensive vascular disease: Report from the PVRI Pediatric Taskforce, Panama 2011. Pulm Circ 2011; 1: 286–298
33) Yoshimura N, Yamaguchi M, Oka S, et al: Inhaled nitric oxide therapy after Fontan type operations. Surg Today 2005; 35: 31e5
34) Miyaji K, Nagata N, Miyamoto T, et al: Combined therapy with inhaled nitric oxide and intravenous epoprostenol (prostacyclin) for critical pulmonary perfusion after the Fontan procedure. J Thorac Cardiovasc Surg 2003; 125: 437e9
35) Agnoletti G, Gala S, Ferroni F, et al: Endothelin inhibitors lower pulmonary vascular resistance and improve functional capacity in patients with Fontan circulation. J Thorac Cardiovasc Surg 2017; 153: 1468–1475 Epub ahead of print
36) Vakilian F, Attaran D, Shegofte M, et al: Assessment of thyroid function in idiopathic pulmonary hypertension. Res Cardiovasc Med 2016; 5: e29361
37) Trapp CM, Elder RW, Gerken AT, et al: Pediatric pulmonary arterial hypertension and hyperthyroidism: A potentially fatal combination. J Clin Endocrinol Metab 2012; 97: 2217–2222
38) Satoh M, Aso K, Nakayama T, et al: Autoimmune thyroid disease in children and adolescents with idiopathic pulmonary arterial hypertension. Circ J 2010; 74: 371–374
39) Yu SC, Chang L, Burke G: Thyrotropin increases prostaglandin levels in isolated thyroid cells. J Clin Invest 1972; 51: 1038–1042
40) Spaulding SW, Burrow GN: Effect of PGE1 and TSH on cAMP dependent protein kinase activity in the thyroid. Endocrinology 1975; 96: 1018–1021
41) Roberts KE, Barst RJ, McElroy JJ, et al: Bone morphogenetic protein receptor 2 mutations in adults and children with idiopathic pulmonary arterial hypertension: Association with thyroid disease. Chest 2005; 128 Suppl: 618S
42) Ivy DD, Doran A, Claussen L, et al: Weaning and discontinuation of epoprostenol in children with idiopathic pulmonary arterial hypertension receiving concomitant bosentan. Am J Cardiol 2004; 93: 943–946
43) Johnson RF, Loyd JE, Mullican AL, et al: Long-term follow-up after conversion from intravenous epoprostenol to oral therapy with bosentan or sildenafil in 13 patients with pulmonary arterial hypertension. J Heart Lung Transplant 2007; 26: 363–369
44) Minai OA, Parambil J, Dweik RA, et al: Impact of switching from epoprostenol to IV treprostinil on treatment satisfaction and quality of life in patients with pulmonary hypertension. Respir Med 2013; 107: 458–465
45) Ivy DD, Claussen L, Doran A: Transition of stable pediatric patients with pulmonary arterial hypertension from intravenous epoprostenol to intravenous treprostinil. Am J Cardiol 2007; 99: 696–698
46) Sitbon O, Manes A, Jais X, et al: Rapid switch from intravenous epoprostenol to intravenous treprostinil in patients with pulmonary arterial hypertension. J Cardiovasc Pharmacol 2007; 49: 1–5
47) Gomberg-Maitland M, Tapson VF, Benza RL, et al: Transition from intravenous epoprostenol to intravenous treprostinil in pulmonary hypertension. Am J Respir Crit Care Med 2005; 172: 1586–1589
48) Simonneau G, Barst RJ, Galie’ N, et al: Treprostinil Study Group: Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: A double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002; 165: 800–804
49) Sadushi-Koliçi R, Skoro-Sajer N, Zimmer D, et al: Long-term treatment, tolerability, and survival with sub-cutaneous treprostinil for severe pulmonary hypertension. J Heart Lung Transplant 2012; 31: 735–743
50) Krishnan U, Takatsuki S, Ivy DD, et al: Effectiveness and safety of inhaled treprostinil for the treatment of pulmonary arterial hypertension in children. Am J Cardiol 2012; 110: 1704–1709
51) Barst RJ, Ivy DD, Gaitan G, et al: A randomized, double-blind, placebo-controlled, dose-ranging study of oral sildenafil citrate in treatment-naive children with pulmonary arterial hypertension. Circulation 2012; 125: 324–334
52) Barst RJ, Beghetti M, Pulido T, et al: STARTS-2 Investigators: STARTS-2: Long-term survival with oral Sildenafil monotherapy in treatment-naive pediatric pulmonary arterial hypertension. Circulation 2014; 129: 1914–1923
53) Galiè N, Brundage BH, Ghofrani HA, et al: Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) Study Group: Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009; 119: 2894–2903
54) Takatsuki S, Calderbank M, Ivy DD: Initial experience with tadalafil in pediatric pulmonary arterial hypertension. Pediatr Cardiol 2012; 33: 683–688
55) Kohno H, Ichida F, Hirono K, et al: Plasma concentrations of tadalafil in children with pulmonary arterial hypertension. Ther Drug Monit 2014; 36: 576–583
56) Sabri MR, Beheshtian E: Comparison of the therapeutic and side effects of tadalafil and sildenafil in children and adolescents with pulmonary arterial hypertension. Pediatr Cardiol 2014; 35: 699–704
57) Ghofrani HA, D’Armini AM, Grimminger F, et al: CHEST-1 Study Group: Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 2013; 369: 319–329
58) Ghofrani HA, Galiè N, Grimminger F, et al: PATENT-1 Study Group: Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013; 369: 330–340
59) Rubin LJ, Galiè N, Grimminger F, et al: Riociguat for the treatment of pulmonary arterial hypertension: A long-term extension study (PATENT-2). Eur Respir J 2015; 45: 1303–1313
60) Galiè N, Müller K, Scalise AV, et al: PATENT PLUS: A blinded, randomised and extension study of riociguat plus sildenafil in pulmonary arterial hypertension. Eur Respir J 2015; 45: 1314–1322
61) Beghetti M, Haworth SG, Bonnet D, et al: Pharmacokinetic and clinical profile of a novel formulation of bosentan in children with pulmonary arterial hypertension: The FUTURE-1 study. Br J Clin Pharmacol 2009; 68: 948–955
62) Berger RM, Haworth SG, Bonnet D, et al: FUTURE-2: Results from an open-label, long-term safety and tolerability extension study using the pediatric FormUlation of bosenTan in pUlmonary arterial hypeRtEnsion. Int J Cardiol 2016; 202: 52–58
63) Hebert A, Mikkelsen UR, Thilen U, et al: Bosentan improves exercise capacity in adolescents and adults after Fontan operation: The TEMPO (Treatment With Endothelin Receptor Antagonist in Fontan Patients, a Randomized, Placebo-Controlled, Double-Blind Study Measuring Peak Oxygen Consumption) study. Circulation 2014; 130: 2021–2030
64) Takatsuki S, Rosenzweig EB, Zuckerman W, et al: Clinical safety, pharmacokinetics, and efficacy of ambrisentan therapy in children with pulmonary arterial hypertension. Pediatr Pulmonol 2013; 48: 27–34
65) Pulido T, Adzerikho I, Channick RN, et al: SERAPHIN Investigators: Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013; 369: 809–818
66) Kerstein D, Levy PS, Hsu DT, et al: Blade balloon atrial septostomy in patients with severe primary pulmonary hypertension. Circulation 1995; 91: 2028–2035
67) Chiu JS, Zuckerman WA, Turner ME, et al: Balloon atrial septostomy in pulmonary arterial hypertension: Effect on survival and associated outcomes. J Heart Lung Transplant 2015; 34: 376–380
68) Kurzyna M, Dabrowski M, Bielecki D, et al: Atrial septostomy in treatment of end-stage right heart failure in patients with pulmonary hypertension. Chest 2007; 131: 977–983
69) Micheletti A, Hislop AA, Lammers A, et al: Role of atrial septostomy in the treatment of children with pulmonary arterial hypertension. Heart 2006; 92: 969–972
70) Blanc J, Vouhé P, Bonnet D: Potts shunt in patients with pulmonary hypertension. N Engl J Med 2004; 350: 623
71) Baruteau AE, Serraf A, Lévy M, et al: Pottsshunt in children with idiopathic pulmonary arterial hypertension: Long-term results. Ann Thorac Surg 2012; 94: 817–824
72) Esch JJ, Shah PB, Cockrill BA, et al: Transcatheter Potts shunt creation in patients with severe pulmonary arterial hypertension: Initial clinical experience. J Heart Lung Transplant 2013; 32: 381–387
73) Hoeper MM, Markevych I, Spiekerkoetter E, et al: Goal oriented treatment and combination therapy for pulmonary arterial hypertension. Eur Respir J 2005; 26: 858–863
74) Sitbon O, Jaïs X, Savale L, et al: Upfront triple combination therapy in pulmonary arterial hypertension. Eur Respir J 2014; 43: 1691–1697
75) Galiè N, Barberà JA, Frost AE, et al: AMBITION Investigators: Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med 2015; 373: 834–844


This page was created on 2017-07-19T17:54:19.194+09:00
This page was last modified on 2017-08-14T17:10:46.589+09:00
このサイトは(株)国際文献社によって運用されています。