肺高血圧症の病態と新規治療をカリウムチャネル制御から探るPotassium Channels in Pulmonary Arterial Hypertension
徳島大学大学院医歯薬学研究部小児科Department of Pediatrics, Institute of Biomedical Science, Tokushima University Graduate School ◇ Tokushima, Japan
発行日:2016年5月1日Published: May 1, 2016
肺動脈平滑筋細胞に存在するカリウムチャネルは多彩な種類と機能を有し,多くの生理作用ならびに病態に関与している.肺動脈性肺高血圧症(PAH)においても発症と増悪に深く関与していることが明らかになりつつある.2013年にtwo-pore domainカリウムチャネルの1種であるKCNK3(TASK1)の遺伝子変異がPAHの原因遺伝子であると証明され,第5回肺高血圧国際シンポジウム(ニース国際会議)で追加された.また,PAHでは発症原因にかかわらず,電位依存性カリウムチャネル,特にKCNA5(Kv1.5)の発現低下と活性抑制が認められ,肺動脈収縮と血管リモデリングを促進することが示されている.さらに,平滑筋細胞に認められるカルシウム活性化カリウムチャネルは,形質転換によってBKca(Kca1.1)からIKca(Kca3.1)優位に変化し,平滑筋細胞の遊走の促進,増殖能の亢進,アポトーシスの抑制などに影響を与えている.これら肺血管の収縮とリモデリングへのカリウムチャネルの役割を解明することで,新たな治療戦略が見えてくるものと期待される.
Potassium channels play diverse roles in regulating the behavior of pulmonary artery smooth muscle cells. In 2013, the discovery of KCNK3 (TASK1) as a new predisposing gene for pulmonary arterial hypertension (PAH) led to an update in the Nice Classification regarding the genetic origin of PAH. Decreased current via KCNA5 (Kv1.5) plays a key role in determining pulmonary arterial tone and vascular remodeling. The transformation of smooth muscle cells causes ion channel switching, such as the loss of BKca (Kca1.1) and the gain of IKca (Kca3.1), in immature proliferative smooth muscle cells and also induces cell migration, proliferation, and apoptosis resistance. Pulmonary smooth muscle cells from PAH patients demonstrate many cellular abnormalities linked to potassium channels. This review summarizes the current knowledge regarding the involvement of potassium channels in the development of PAH and discusses potential treatments to be developed in the near feature.
Key words: potassium channel; pulmonary arterial hypertension; vascular
© 2016 特定非営利活動法人日本小児循環器学会© 2016 Japanese Society of Pediatric Cardiology and Cardiac Surgery
肺高血圧症は様々な原因により肺動脈圧,肺血管抵抗が持続的に上昇した病態であり,右心不全や呼吸不全が進行性に悪化する予後不良の難治性疾患である.肺動脈性肺高血圧症(pulmonary arterial hypertension: PAH)の病態の主体は肺動脈内腔の狭窄であり,①血管拡張因子と血管収縮因子のアンバランスなどによる直径500 µm以下の末梢の肺小動脈の異常収縮,②血管内皮細胞および平滑筋細胞などの過剰増殖とアポトーシス抵抗性による血管リモデリング,③病変部での血栓形成,の3つの要因によって生じる.病初期には異常収縮が大部分を占め,徐々に血管リモデリングの比率が大きくなる.このような病態には肺動脈の内皮細胞および平滑筋細胞の特性が関与している1).PAHの病変は血管壁の肥厚により内腔が狭窄・閉塞してくる収縮性病変と叢状病変,拡張性病変,血管炎からなる複合病変に大別される.病初期には,孤立性中膜肥厚を示すが,肺高血圧が続くと内膜の肥厚が起こってくる2, 3).主に平滑筋細胞,筋線維芽細胞など細胞成分の増加によるものを細胞性内膜肥厚,膠原線維を主とする線維成分の増生によるものを線維性内膜肥厚と呼ぶ.
本疾患の原因遺伝子として,bone morphogenic protein type II receptor遺伝子(BMPR2),Activin receptor-like kinase-1(ALK-1)遺伝子(ACVRL1),endogolin遺伝子(ENG),SMAD8/9(SMAD9)遺伝子などのTGF-βシグナル関連遺伝子に加えて,細胞内カルシウム調節因子であるCaveolin-1(CAV1)遺伝子,そして2013年にはカリウムチャネルであるKCNK3(TASK1)遺伝子の変異が証明され4),第5回肺高血圧国際シンポジウム(ニース)で追加された5).このカリウムチャネル遺伝子変異の発見は,TGF-βシグナル伝達に関連しないPAH発症機序に関する新たな知見につながる可能性がある.PAHの発症と増悪におけるカリウムチャネルの関与については,電位依存性カリウムチャネルの一つであるKCNA5(Kv1.5)が取り上げられることが多かった6, 7).Kv1.5電流の低下は静止膜電位を浅くし,肺血管を収縮させるのみでなく,細胞増殖や遊走にも影響を与える.さらに細胞内カリウムイオン濃度の増加によりカスパーゼ活性が抑制され,アポトーシス抵抗性を誘導する作用も有している.また,KCNK3,KCNA5以外にも,多くのカリウムチャネルは肺小動脈の収縮・弛緩およびリモデリング,そして肺動脈平滑筋細胞の増殖,アポトーシスおよび遊走に関与しており,今後の治療戦略に大きな位置を占める可能性がある.現在,プロスタサイクリン製剤,エンドセリン受容体拮抗薬,ホスホジエステラーゼ-5(PDE-5)阻害薬の使用が広く普及し,ランダム化比較対象臨床試験を含む単剤治療および併用療法の成績が集積されてきているが8–10),難治性である本疾患の治療成績はいまだ満足できるものではなく,さらなるbreakthroughが必要であろう11).この総説では肺動脈性肺高血圧症におけるカリウムチャネルの働きと制御に関して概要を述べ,将来的な治療戦略への展望について解説する.
カリウムチャネルはカリウムイオンを選択的に透過させる細胞膜に存在するイオンチャネルである.静止膜電位の形成,細胞の興奮性,電気的な細胞応答,活動電位の形成と持続時間,シナプス伝達,細胞分裂,細胞分化,周期性活動,張力など種々の生体の調整や細胞の機能制御に重要な役割を演じている.血管平滑筋細胞に存在するカリウムチャネルは,電位依存性カリウムチャネル(voltage-gated K+ channel; Kv),カルシウム活性化カリウムチャネル(Ca2+-activated K+ channel; Kca),Two-pore domainカリウムチャネル(Two-pore domain K+ channel; K2P),内向き整流性カリウムチャネル(Inwardly rectifying K+ channel; KIR)の4つに大きく分類される12, 13)(Table 1).ほとんどのカリウムチャネルはαサブユニットが四量体を形成し,中央部分にカリウムを通す小孔(ポア)が開くようになっている.電気生理学的特性やαサブユニットの膜貫通領域の構造の違いにより,6回膜貫通型のKv,Kca,2回膜貫通型のKIR,4回膜貫通型のK2Pに大別される.イオン透過経路を構成するαサブユニットと電流特性や膜発現量を制御するβサブユニットをあわせると100種類以上の遺伝子群から構成されており,これら豊富なサブユニット分子種,αサブユニットのヘテロ四量体形成,さらにβサブユニットとの複合体形成によってカリウムチャネルの多様性と多彩な機能が発現する.
| K+ channel Families | Kv channel (42 isoforms; 12 subfamilies) | KCa channel (8 isoforms; 5 subfamilies) | K2P channel (15 isoforms; 6 subfamilies) | KIR channel (15 isoforms; 7 subfamilies) | ||||
|---|---|---|---|---|---|---|---|---|
| Nomenclature | HGCN | IUPHAR | HGCN | IUPHAR | HGCN | IUPHAR | HGCN | IUPHAR |
| Isoforms & Subfamilies | KCNA1–A10 | Kv1.1–Kv1.10 | KCNMA1 | KCa1.1 | KCNK1 | K2P1.1 | KCNJ1 | KIR1.1 |
| KCNB1 & B2 | Kv2.1 & Kv2.2 | KCNN1–3 | KCa2.1–2.3 | KCNK2 | K2P2.1 | KCNJ2, 12, 4, 14 | KIR2.1–KIR2.4 | |
| KCNC1–C4 | Kv3.1–Kv3.4 | KCNN4 | KCa3.1 | KCNK3 | K2P3.1 | KCNJ3, 6, 9. 5 | KIR3.1–KIR3.4 | |
| KCND1–D3 | Kv4.1–Kv4.3 | KCNT1 & T2 | KCa4.1 & 4.2 | KCNK4 | K2P4.1 | KCNJ10 & 15 | KIR5.1 | |
| KCNF1 | Kv5.1 | KCNU1 | KCa5.1 | KCNK5 | K2P5.1 | KCNJ8 & 11 | KIR6.1 & 6.2 | |
| KCNG1–G4 | Kv6.1–Kv6.4 | KCNK6 | K2P6.1 | KCNJ13 | KIR7.1 | |||
| KCNQ1–Q5 | Kv7.1–Kv7.5 | KCNK7 | K2P7.1 | |||||
| KCNV1 & V2 | Kv8.1 & Kv8.2 | KCNK9 | K2P9.1 | |||||
| KCNS1–3 | Kv9.1–Kv9.3 | KCNK10 | K2P10.1 | |||||
| KCNH1 & 5 | Kv10.1 & Kv10.2 | KCNK12 | K2P12.1 | |||||
| KCNH2, H6, H7 | Kv11.1–Kv11.3 | KCNK13 | K2P13.1 | |||||
| KCNH8, H3, H4 | Kv12.1–Kv12.3 | KCNK15 | K2P15.1 | |||||
| KCNK16 | K2P16.1 | |||||||
| KCNK17 | K2P17.1 | |||||||
| KCNK18 | K2P18.1 | |||||||
| General characteristic of the K+ channels | Ca2+-insensitive, voltage-sensitive | KCa1.1 (BKca, Slo1, or MaxiK): large-conductance, voltage- and Ca2+-sensitive, outward rectification | Voltage-independent | Single-channel conductance: <30 pS and inward rectification | ||||
| Outward rectification | Single-channel conductance: ≈250 pS | Single-channel conductance: <40pS, except TREK (≈100 pS) | KIR subfamily: strong inward rectification | |||||
| Single-channel conductance: 5–80 pS | KCa2.1–2.3 (SKca), KCa3.1 (IKca): small- and intermediate-conductance, and voltage independent | TWIK, TREK, TASK, TALK, THINK, TRAAK subfamilies | KATP subfamily (KIR6.1 & 6.2): intermediate inward rectification, intracellular ATP sensitive | |||||
| Single-channel conductance: SKca 10–14 pS, IKca ≈45 pS | ||||||||
| α-subunit membrane topology | 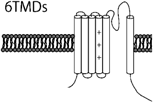 | 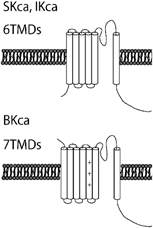 | 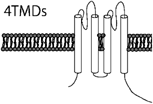 | 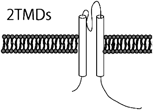 | ||||
| Human potassium channels can be broken down into 4 distinct families by their functional characteristics. Kv, voltage-gated; KCa, calcium activated; K2P, two pore; KIR, inward rectifying; HGCN, HUGO human genome organization nomenclature; IUPHAR, International Union of Pharmacology nomenclature; TMDs, transmebrane domains; +, voltage sensor. | ||||||||
肺動脈は様々な血管作動性物質や環境・ストレス・薬剤によって収縮と拡張を制御されている.これらのなかでも肺血管に特徴的な応答である低酸素による血管収縮について示した(Fig. 1)14, 15).正常酸素分圧下においては血管平滑筋細胞の膜電位は,−50~−60 mVで維持されており,カルシウムイオンの電位依存性カルシウムチャネル(voltage-dependent Ca2+ channel; VDCC)からの流入が抑制されている.
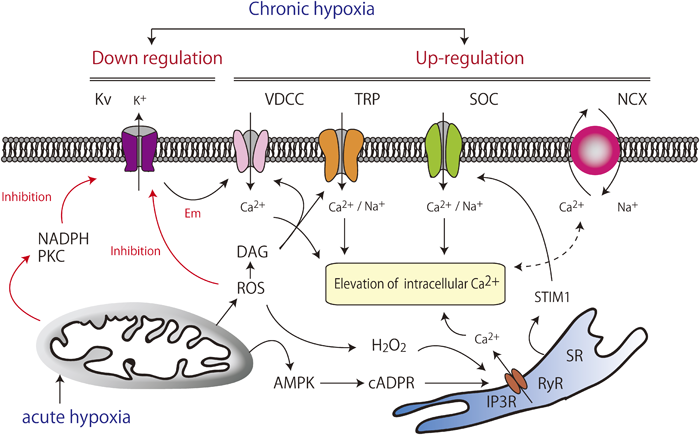
Vasoconstriction involves hypoxia-induced elevation of intracellular Ca2+ and the related signaling pathways. The inhibition of Kv channels, particularly Kv1.5, plays a key role in the mechanism of vasoconstriction. AMPK, AMP-activated kinase; cADPR, cyclic ADP ribose; DAG, diacylglycerol; Em, membrane potential; IP3R, Inositol 1,4,5-trisphosphate receptor; Kv, voltage-gated K+ channels; NCX, Na+–Ca2+ exchanger; PKC, protein kinase C; ROS, reactive oxygen species; RyR, ryanodine receptor; SOC, store-operated channels; SR, sarcoplasmic reticulum; STIM1, stromal-interacting molecule 1; TRP, transient receptor potential channels; VDCC, voltage-dependent Ca2+ channels.
カリウムチャネルの発現低下や活性の抑制は平滑筋細胞におけるカリウム電流低下を来たし,静止膜電位の上昇・脱分極をもたらす.これによってVDCCが活性化し,細胞内カルシウム濃度を上昇させて筋原性張力が発生し,血管平滑筋を収縮させることがシグナル伝達経路として確立している16, 17).このカルシウム増加は筋小胞体(sarcoplasmic reticulum)からのCa2+放出を促す(カルシウム誘発性カルシウム放出:Calcium-induced calcium release; CICR)作用にもつながる.
膜電位を介した血管平滑筋細胞収縮の調節系においては,BKcaがフィードバック機構の要である.細胞内カルシウム濃度が上昇するほど,また脱分極するほどBKcaが活性化される特性はフィードバック機構に適している.BKcaが活性化することによって過分極がもたらされ,VDCCをはじめとする電位依存性チャネルの抑制を促すのである.Kvチャネルも脱分極により開放する性質があるため負のフィードバックに大きく関与している.
細胞膜電位の脱分極や持続する細胞内カルシウム濃度の上昇は,生理的にも病的にも,平滑筋細胞の収縮だけでなく,細胞増殖を刺激する大きな要因である.核内および細胞質のカルシウム濃度の上昇により,calmodulin kinase(CaMK)やmitogen-activated protein kinase(MAPK)などのCa2+依存性キナーゼが,さらにnuclear factor of activated T-cells(NFAT)やcAMP response element binding protein(CREB)などの転写因子が活性化され,静止期にある細胞が細胞周期に入り増殖が促される(Fig. 2)16).この細胞シグナル伝達制御に,Kvなどのカリウムチャネルが関わっている.
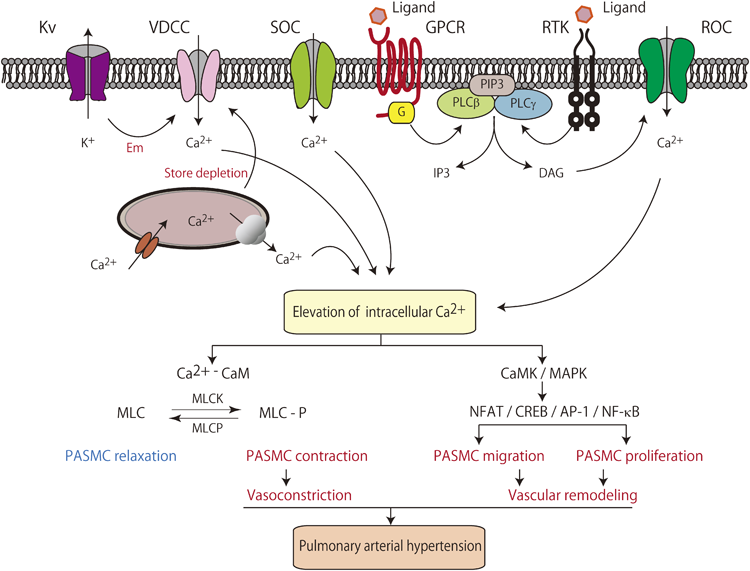
A rise in cytosolic Ca2+ can be created by opening voltage-dependent Ca2+ channels (VDCC) through decreased voltage-gated K+ (KV) channel current and membrane depolarization (Em). Activation of receptors, such as G protein-coupled receptors (GPCR) and receptor tyrosine kinases (RTK), induces diacylglycerol (DAG) and inositol 1,4,5,-trisphosphate (IP3) production. In addition, these receptors increase the cytosolic Ca2+ concentration by opening receptor-operated Ca2+ channels (ROC) and inducing Ca2+ mobilization from the sarcoplasmic reticulum (SR). IP3 also directly or indirectly opens store-operated Ca2+ channels (SOC) by store depletion to further increase Ca2+. The Ca2+/calmodulin (CaM) complex binds to and activates myosin light chain kinase (MLCK), which phosphorylates the myosin light chain (MLC). MLC stimulates the activity of myosin adenosine triphosphatase (ATPase), which hydrolyzes ATP to generate energy for cycling of myosin cross-bridges with actin filaments. Formation of these cross-bridges underlies pulmonary artery smooth muscle cell (PASMC) contraction, prompting vasoconstriction. Furthermore, an elevation in the intracellular Ca2+ concentration induces quiescent cells to undergo mitosis. Increased intracellular Ca2+ also activates CaM kinase (CaMK) and mitogen-activated protein kinase (MAPK), as well as transcription factors, including nuclear factor of activated T cells (NFAT), cAMP response element binding protein (CREB), activator protein-1 (AP-1), and NF-κB, to stimulate proliferation by inducing Ca2+-sensitive steps during cell cycle progression. Chronic and sustained elevation of pulmonary vascular resistance and arterial pressure resulted from vasoconstriction and vascular remodeling.
KvチャネルのなかでもKv1.5(KCNA5)は弾性動脈・筋性動脈よりも細小動脈に多く発現しており,急性期には低酸素によってKv1.5活性の抑制がもたらされ,カリウム電流低下,細胞膜の脱分極を介して平滑筋の収縮が起こる.低酸素状態により活性酸素(reactive oxygen species; ROS)増加,ニコチンアミドアデニンジヌクレオチドリン酸(nicotinamide adenine dinucleotide phosphate; NADPH)増加,スフィンゴミエリナーゼ活性化によるProtein kinase C(PKC)の活性化が起こる.これらのシグナルはすべて,急性期にはKv1.5活性を抑制する.さらに肺胞低酸素は肺血管収縮を引き起こすのみでなく,慢性的には肺小動脈におけるKv1.5の発現を抑制してリモデリングを促進する18).Kv1.5の発現減少は肺高血圧の原因にかかわらず,共通の性質・特性として認められており,病態の増悪には非常に重要と考えられている.この原因は明らかにされていないが,多因子の関与が示唆され,今後の治療のターゲットとなり得る.
2013年にPAH原因遺伝子として報告されたKCNK3(TASK1)は,Two-pore domainカリウムチャネル(K2P)の1種である.K2Pは2回膜貫通領域と一つのP領域が2個直列につながったサブユニット構造をしており,低酸素やpHを感知して活性化される.電気生理学的特性や薬理学的な特性から6つのサブファミリー(TWIK,TREK,TASK,TALK,THINK,TRAAK)に分類されている.この報告では6つのヘテロ接合性ミスセンスバリアントが独立に同定された4).KCNK3は電位依存性チャネルではなく静止膜電位付近でも活性を示すため,このチャネル異常は膜電位維持を阻害することで肺血管収縮とリモデリングを促進すると考えられる.
一方では,多くの臓器において低酸素や虚血などで活性化されるATP-sensitive K+ channel(KATP)19, 20)については肺動脈における収縮とリモデリングやフィードバックへの関与の報告は少ない21).特に細胞膜KATP channelは病態への関与において重要性に乏しいと考えられる.ミトコンドリアKATP channelに関しては肺動脈収縮やリモデリングへの影響を示唆する報告も散見されるが21),病態増悪における役割はいまだ明確には示されていない.
一般に終末分化した細胞(骨格筋細胞・神経細胞・血球系細胞など)は,その分化形質を細胞死に至るまで維持し,脱分化や細胞分裂などを示さない,いわゆる最終分化(terminal differentiation)を示す.一方,平滑筋細胞は,病的あるいは特殊な条件下(肺高血圧・動脈硬化・病的血管・培養など)において容易に分化型形質から脱分化型形質へと変化する.また,分化型形質を維持したまま細胞増殖性も認めるなど,特有の可塑性を示している.
血管平滑筋細胞の形質転換(脱分化)が起点となり,脱分化型血管平滑筋細胞の増殖・遊走によって血管内中膜壁肥厚などが引き起こされ,血管壁リモデリングが増悪する.近年,血管平滑筋細胞の形質転換には種々のイオンチャネルの発現増加・減少が密接にかかわっていることが明らかにされつつある22, 23).平滑筋細胞が増殖停止し分化した状態では,興奮収縮連関に関与するVDCCやBKcaの発現が優位である.これらのチャネルは,前述のように細胞膜電位に依存して平滑筋細胞のカルシウム流入の重要な制御因子として働く24).これに対して血管平滑筋細胞に増殖刺激などが加わると,VDCCやBKcaの発現が急速に減少する25).代わって,transient receptor potential channel(TRP)やIntermediate-conductance Ca2+-activated K+ channel(IKca; Kca3.1)の発現が増加することが明らかにされた(Fig. 3)23, 25).分化型平滑筋細胞に発現しているVDCCおよびBKcaは膜電位に強く依存して活性化されるのに対して,TRPやIKcaは膜電位にほとんど影響されずに静止膜電位付近でも活性化して開口する特性を持っている.したがって増殖型刺激が加わり脱分化型(増殖型)に転じた血管平滑筋細胞では,IKcaの活性化による過分極作用によって細胞膜電位が過分極され,電位非依存的経路であるTRPを介した恒常的なカルシウム流入を駆動するための電位差が大きく保たれることとなるのである22, 23).TRPを介した細胞内カルシウム濃度の上昇はさらにIKcaを活性化させ,ポジティブ・フィードバックを示す.このような状態は,前述したNFAT/CREB/AP-1/NF-κBなどの細胞内カルシウム濃度依存性転写因子の活性化を起こすのに好都合である.NFATやNF-κBには,TRPやIKcaの発現を増加させる作用も認められ,さらにポジティブ・フィードバックを受けて拍車がかかることになる25).
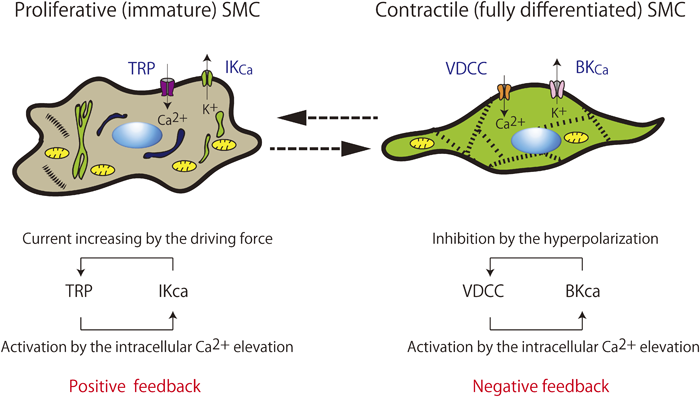
Vascular smooth muscle cells (SMCs) can have one of the two phenotypes: immature proliferative SMCs and differentiated contractile SMCs. Vascular SMCs change phenotype in response to the surrounding environment. Immature proliferative SMCs proliferate, migrate, and synthesize proteins. In contrast, fully differentiated contractile SMCs adhere to each other and are contractile in nature. Switching to different ion transport systems is also shown. This phenotypic shift in the Ca2+-activated K+ channel (Kca) expression pattern produces dramatic alterations in the electrical properties of the cell and has functional consequences, in part because of the effect on Ca2+ influx. Activation of IKca enhances Ca2+ influx by increasing the transmembrane electrical gradient. This increase in Ca2+ influx stimulates distinct cellular processes associated with smooth muscle growth and proliferation. BKca, large conductance Ca2+-activated K+ channel; IKca, intermediate conductance Ca2+-activated K+ channel; SMC, smooth muscle cell; TRP, transient receptor potential channels; VDCC, voltage-dependent Ca2+ channels.
Fig. 4は,幼若な増殖型平滑筋細胞におけるIKcaの発現増加を,patch-clamp法(whole-cell configurationおよびinside-out patch)を用いて証明した記録である.増殖型平滑筋細胞のwhole-cell configurationにおけるカリウム電流は,BKca選択的阻害薬であるIberiotoxin(IbTX)投与では軽度(14%)しか抑制されなかった.その後に追加したBKcaおよびIKca阻害薬であるcharybdotoxin(ChTX)によって,カリウム電流が強く抑制された(Fig. 4B).分化型平滑筋細胞ではIKcaはほとんど発現しておらず,IKcaを介した電流もごくわずかであるとされているが,増殖型平滑筋細胞ではIKca電流が著しく増加していることを示している.さらに,Inside-out patchを用いたsingle channel recordingでは,チャネルコンダクタンス,電位非依存性およびカルシウム依存性チャネル活性などの電気生理学的性質から細胞膜に存在するチャネルがIKcaであることが同定された(Fig. 4D–F).これらの結果から,増殖型平滑筋細胞のカルシウム依存性カリウム電流の大部分がIKcaを介した電流であり,IKcaの発現が増加していることが確認された.Transient receptor potential(TRP)チャネルは6回膜貫通型の4量体チャネルで,ナトリウム,カリウム,カルシウムなどを透過する非選択的陽イオンチャネルである.膜電位センサー様の構造を持つが,膜電位感受性は非常に弱いか認められず,生体内外からの様々な環境刺激や細胞内外のシグナル,リガンドで活性化される.心肥大や冠状動脈のリモデリングへの関与で注目されるTRPC1よりも,TRPC6によるストア作動性カルシウム流入チャネル活性増加が肺動脈平滑筋細胞の増殖や異常に関わると考えられている.PAHの患者から採取した平滑筋細胞では,TRPC6の発現増加に伴うストア作動性カルシウムチャネル活性増加と増殖能亢進がみられる.これらの増殖性変化は,in vitroではsiRNAによる発現抑制によって阻害されることが証明されている26).
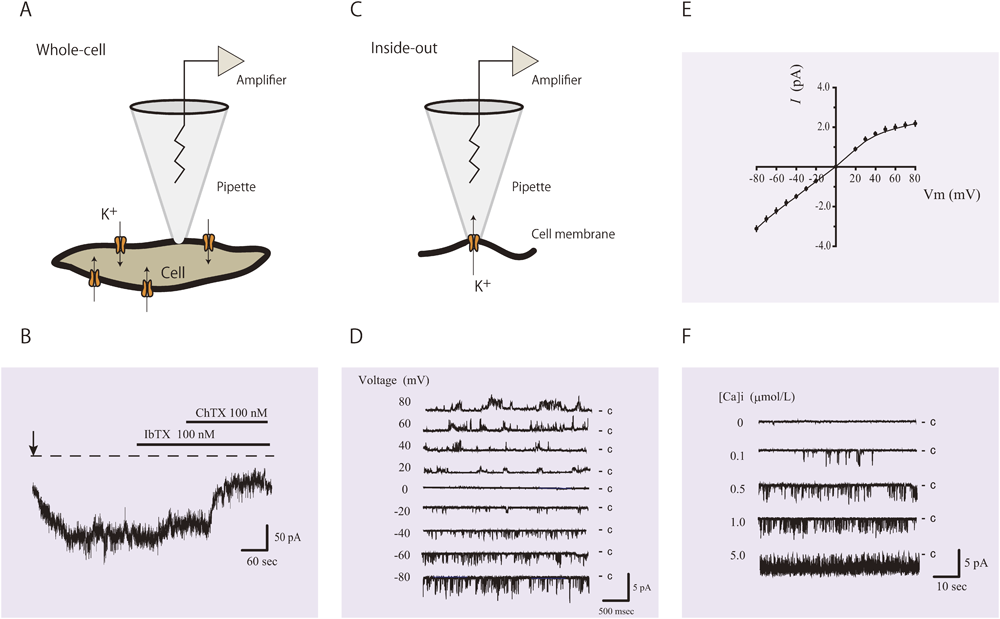
IKca current is observed using the patch-clamp technique. (A) Schematic of the whole-cell configuration. (B) Representative recording of whole-cell current from an immature SMC held at −60 mV. The current shows iberiotoxin (IbTX)-insensitive and charybdotoxin (ChTX)-sensitive K+ currents. Establishment of the whole-cell configuration is shown by the vertical arrow. The zero current level is shown by the dashed line. IbTX inhibited the ChTX-sensitive currents by 14% in this cell. (C) Schematic of an inside-out patch. (D) IKca single-channel currents from an inside-out patch. The membrane potential was stepped from −80 to +80 mV in 10-mV increments. (E) Single-channel current–voltage (I–V) curve obtained in symmetrical K+ solutions, depicting a channel conductance of 37 pS. (F) Intracellular Ca2+ activation of IKca current. Representative traces show single-channel activity of inside-out patches exposed to 0, 0.1, 0.5, 1.0, and 5.0 µmol/L free Ca2+. These recordings are from our experimental data (reference 23).
血管リモデリングの進展には,平滑筋細胞の増殖とともに遊走も大きな要因となる27).細胞遊走の基本は,細胞の前方が伸展・突出し,後方が収縮・短縮することの繰り返しであり,細胞前方の容積増加と後方の容積減少が必要である.細胞容積の増減は特にカリウムチャネルなどのイオンチャネルやトランスポーターによって制御されており,さらに細胞骨格・アクチンフィラメントなどとも協同的に形成されている.まず細胞内前方のCl−/HCO3− exchanger(AE2),Na+/H+ exchanger(NHE)などが活性化され,塩類移動と浸透圧変化に伴う水分の細胞内流入が細胞容積膨張を惹起させる.細胞容積が膨らむことで細胞膜が伸展し,機械受容(伸展活性化)チャネルが活性化され,細胞内にカルシウムが流入する28, 29).この細胞内カルシウム濃度の上昇が細胞後方に存在するIKcaを活性化し,カリウムが細胞外へ流出することに伴い,細胞後方が縮小することとなる(Fig. 5)27).この繰り返しによって遊走が成立し,この作用においても脱分化した平滑筋細胞に多く発現するIKcaが重要な役割を果たしている.
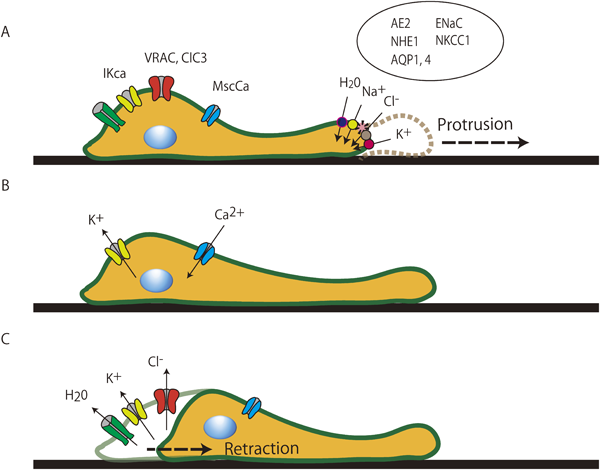
As shown in the schematic, cell migration is a continuous cycle of protrusion of the cell front followed by the retraction of the trailing end. This process can be represented as a cycle of isosmotic volume increases at the cell front and isosmotic volume decreases at the rear end. Extension of the lamellipodium results from salt and osmotically obliged water uptake mediated by the parallel operation of Na+/H+ and Cl−/HCO3− exchange as well as Na+-HCO3− cotransport at the front of migrating cells. Increases in volume and membrane tension eventually produce an increase in the intracellular Ca2+ concentration via activation of Ca2+-permeable stretch-activated cation channels. The rise in intracellular Ca2+ concentration induces retraction of the rear portion of the migrating cell, which is paralleled by massive K+ efflux and shrinkage of the cell pole. AE2, Cl−/HCO3− exchanger isoform 2; AQP 1, 4, aquaporin 1, 4; ClC3, ClC3 chloride channel; ENaC, epithelial Na+ channel; IKca, intermediate conductance Ca2+-activated K+ channel; MScCa, mechanosensitive cation channel; NHE1, Na+/H+ exchanger isoform 1; NKCC1, Na+/K+/2Cl− cotransporter isoform 1; VRAC, volume-regulated anion channels.
細胞増殖とアポトーシスは,正常組織を維持するための相反する制御である.アポトーシスでは,形態的,生化学的変化による細胞容積縮小が最初に認められる.これは,K+, Cl−, H2Oが細胞外へ流出することによって生じる.この後,核凝縮とDNA断片化が起こる.カリウムチャネル活性化に伴うカリウムの細胞外流出は,細胞容積縮小に対して重要なだけでなく,カスパーゼ活性化やDNA断片化にも大きな役割を担っているとされている.肺高血圧症における平滑筋細胞のアポトーシス抵抗性には,Kv1.5,mitochondrial KATP, mitochondrial BKcaの関与が示唆されている30).
現在までPAHの治療は,プロスタサイクリン製剤,エンドセリン受容体拮抗薬,ホスホジエステラーゼ-5(PDE-5)阻害薬などの肺動脈収縮と拡張に関するシグナル伝達経路に焦点があてられている.これらの治療薬は確かにPAHの治療成績を飛躍的に向上させたが,限界もあり,肺動脈壁のリモデリング制御の研究が注目されつつある.肺動脈平滑筋細胞のカリウムチャネルは,治療標的の一つとして有力な候補と考えられる.たとえば,近年,遺伝性PAHの新たな原因遺伝子として特定されたKCNK3の報告4)においては,KCNK3の電気生理学的検討により同遺伝子のミスセンスバリアントはすべて機能喪失をもたらすことが示された.このKCNK3電流の減少はホスホリパーゼ阻害薬(ONO-RS-082)の投与により改善されたと報告されている.本論文においては原因遺伝子特定以上に特異的治療薬の同定の意義も非常に大きく,薬理学的操作で改善を認めていることは重要である.また,低酸素暴露による肺高血圧ラットに対してSurvivin阻害薬であるYM155を投与することによってKvチャネルの発現増加と活性化が認められるとの報告がある31).Kv channelの抑制はPAH症例の原因疾患にかかわらず認められるものであり,応用されれば幅広い症例に効果が得られる可能性がある.さらに,遊走,増殖,形質転換の際に中心的な役割を担うIKcaの阻害薬であるTRAM-34は,血管リモデリング抑制に関するデータが認められており32),将来的な臨床応用が期待される.
上記に示したような薬理学的な手法だけではなく,PAHでは発現が低下してしまっているKCNK3やKCNA5を肺動脈平滑筋細胞に遺伝子導入することなどは将来的な治療方法となる可能性がある.今後,カリウムチャネル制御の解明はPAH治療において大きなインパクトとなると考えられる.
本総説ではカリウムチャネルが様々な場面において肺高血圧症に関与していることを示した.肺血管収縮およびリモデリングへのカリウムチャネルの役割を解明することで肺動脈性肺高血圧症に対する新たな治療戦略が確立されるものと期待される.
1) 早渕康信:肺高血圧症:主に肺動脈性肺高血圧症について.小児内科46巻増刊号小児疾患診療のための病態生理1 改訂5版.東京医学社,2014, pp 347–352
2) Tuder RM, Abman SH, Braun T, et al: Development and pathology of pulmonary hypertension. J Am Coll Cardiol 2009; 30 Suppl: S3–S9
3) 早渕康信,阪田美穂,小野朱美,ほか:光干渉断層像(Optical Coherence Tomography)で肺血管病変を観察した特発性肺動脈性肺高血圧症.日小児循環器会誌2014; 30: 365–370
4) Ma L, Roman-Campos D, Austin ED, et al: A novel channelopathy in pulmonary arterial hypertension. N Engl J Med 2013; 369: 351–361
5) Simonneau G, Gatzoulis MA, Adatia I, et al: Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62 Suppl: D34–D41
6) Archer SL, Wu XC, Thébaud B, et al: Preferential expression and function of voltage-gated, O2-sensitive K+ channels in resistance pulmonary arteries explains regional heterogeneity in hypoxic pulmonary vasoconstriction: Ionic diversity in smooth muscle cells. Circ Res 2004; 95: 308–318
7) Wang J, Juhaszova M, Rubin LJ, et al: Hypoxia inhibits gene expression of voltage-gated K+ channel alphasubunits in pulmonary artery smooth muscle cells. J Clin Invest 1997; 100: 2347–2353
8) Macchia A, Marchioli R, Tognoni G, et al: Systematic review of trials using vasodilators in pulmonary arterial hypertension: Why a new approach is needed. Am Heart J 2010; 159: 245–257
9) Galiè N, Palazzini M, Manes A: Pulmonary arterial hypertension: From the kingdom of the near-dead to multiple clinical trial meta-analyses. Eur Heart J 2010; 31: 2080–2086
10) Fox BD, Shimony A, Langleben D: Meta-analysis of monotherapy versus combination therapy for pulmonary arterial hypertension. Am J Cardiol 2011; 108: 1177–1182
11) 早渕康信:一酸化窒素吸入療法とPhosphodiesterase-5阻害薬による肺高血圧および心不全治療の可能性.日小児循環器会誌2014; 30: 36–38
12) Bonnet S, Archer SL: Potassium channel diversity in the pulmonary arteries and pulmonary veins: Implications for regulation of the pulmonary vasculature in health and during pulmonary hypertension. Pharmacol Ther 2007; 115: 56–69
13) González C, Baez-Nieto D, Valencia I, et al: K(+) channels: Function-structural overview. Compr Physiol 2012; 2: 2087–2149
14) Ward JP, McMurtry IF: Mechanisms of hypoxic pulmonary vasoconstriction and their roles in pulmonary hypertension: New findings for an old problem. Curr Opin Pharmacol 2009; 9: 287–296
15) Stenmark KR, Fagan KA, Frid MG: Hypoxia-induced pulmonary vascular remodeling: Cellular and molecular mechanisms. Circ Res 2006; 99: 675–691
16) Kuhr FK, Smith KA, Song MY, et al: New mechanisms of pulmonary arterial hypertension: Role of Ca2+ signaling. Am J Physiol Heart Circ Physiol 2012; 302: H1546–H1562
17) Hayabuchi Y, Standen NB, Davies NW: Angiotensin II inhibits and alters kinetics of voltage-gated K(+) channels of rat arterial smooth muscle. Am J Physiol Heart Circ Physiol 2001; 281: H2480–H2489
18) Burg ED, Remillard CV, Yuan JX: Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: Pharmacotherapeutic implications. Br J Pharmacol 2008; 153 Suppl 1: S99–S111
19) Hayabuchi Y, Willars GB, Standen NB, et al: Insulin-like growth factor-I inhibits rat arterial K(ATP) channels through pI 3-kinase. Biochem Biophys Res Commun 2008; 374: 742–746
20) Hayabuchi Y, Dart C, Standen NB: Evidence for involvement of A-kinase anchoring protein in activation of rat arterial K(ATP) channels by protein kinase A. J Physiol 2001; 536: 421–427
21) Sahara M, Sata M, Morita T, et al: Nicorandil attenuates monocrotaline-induced vascular endothelial damage and pulmonary arterial hypertension. PLoS ONE 2012; 7: e33367
22) Landsberg JW, Yuan JX: Calcium and TRP channels in pulmonary vascular smooth muscle cell proliferation. News Physiol Sci 2004; 19: 44–50
23) Hayabuchi Y, Nakaya Y, Yasui S, et al: Angiotensin II activates intermediate-conductance Ca2+-activated K+ channels in arterial smooth muscle cells. J Mol Cell Cardiol 2006; 41: 972–979
24) Hayabuchi Y, Nakaya Y, Matsuoka S, et al: Endothelium-derived hyperpolarizing factor activates Ca2+-activated K+ channels in porcine coronary artery smooth muscle cells. J Cardiovasc Pharmacol 1998; 32: 642–649
25) Beech DJ: Ion channel switching and activation in smooth-muscle cells of occlusive vascular diseases. Biochem Soc Trans 2007; 35: 890–894
26) Yu Y, Sweeney M, Zhang S, et al: PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol 2003; 284: C316–C330
27) Schwab A, Fabian A, Hanley PJ, et al: Role of ion channels and transporters in cell migration. Physiol Rev 2012; 92: 1865–1913
28) Hayabuchi Y, Sakata M, Ohnishi T, et al: Mechanical stretch and Intermediate-conductance Ca2+-activated K+ channels in arterial smooth muscle cells, in Kamkin A, Lozinsky I (eds): Mechanosensitivity in Cells and Tissues, Vol. 6. Mechanically gated channels and their regulation, 2012
29) Hayabuchi Y, Nakaya Y, Mawatari K, et al: Cell membrane stretch activates intermediate-conductance Ca2+-activated K+ channels in arterial smooth muscle cells. Heart Vessels 2011; 26: 91–100
30) Remillard CV, Yuan JX: Activation of K+ channels: An essential pathway in programmed cell death. Am J Physiol Lung Cell Mol Physiol 2004; 286: L49–L67
31) Fan Z, Liu B, Zhang S, et al: YM155, a selective survivin inhibitor, reverses chronic hypoxic pulmonary hypertension in rats via upregulating voltage-gated potassium channels. Clin Exp Hypertens 2015; 37: 381–387
32) Wulff H, Castle NA: Therapeutic potential of KCa3.1 blockers: Recent advances and promising trends. Expert Rev Clin Pharmacol 2010; 3: 385–396


This page was created on 2016-05-11T16:50:49.498+09:00
This page was last modified on 2016-05-24T16:08:55.677+09:00
このサイトは(株)国際文献社によって運用されています。