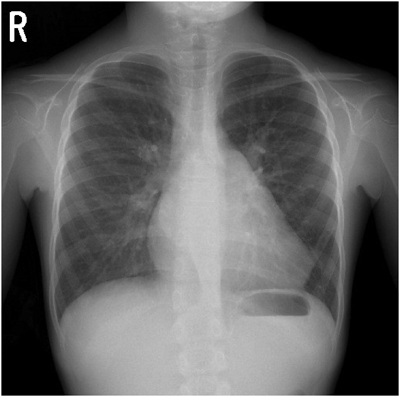
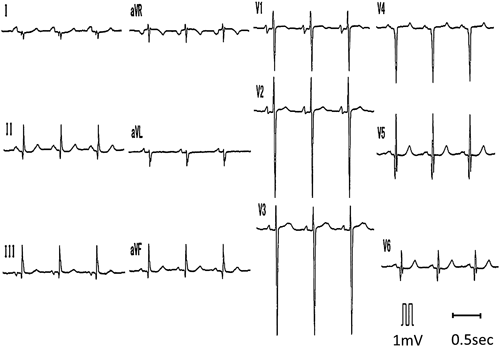
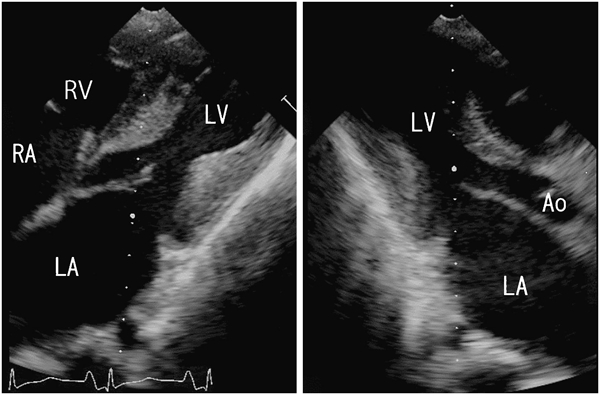
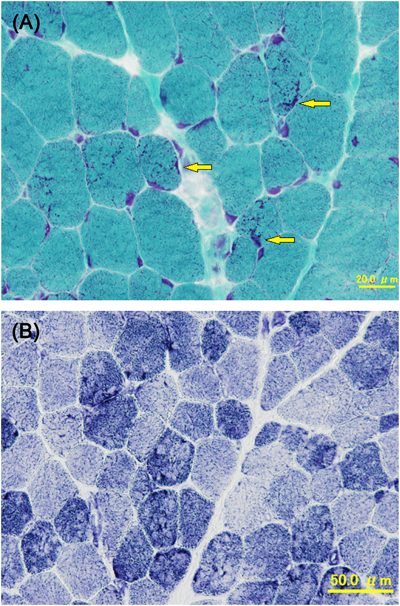
ACTA1変異を伴うネマリンミオパチーに合併した肥大型心筋症の1例Hypertrophic Cardiomyopathy Associated with Nemaline Myopathy Due to ACTA1 Mutation
1 東京都立小児総合医療センター循環器科Department of Cardiology, Tokyo Metropolitan Children’s Medical Center ◇ Tokyo, Japan
2 東京都立小児総合医療センター神経内科Department of Neurology, Tokyo Metropolitan Children’s Medical Center ◇ Tokyo, Japan
3 国立精神・神経医療研究センター神経研究所疾病研究第一部Department of Neuromuscular Research, National Center of Neurology and Psychiatry ◇ Tokyo, Japan
4 東京医科大学病態生理学分野Department of Pathophysiology, Tokyo Medical University ◇ Tokyo, Japan
受付日:2015年10月31日Received: October 31, 2015
受理日:2015年12月24日Accepted: December 24, 2015
発行日:2016年3月1日Published: March 1, 2016